
Enzymatic Degradation Studies: Assessing Peptide Stability
Author: Dr. Numan S. Date: October 9, 2025

What Is Enzymatic Degradation?
Enzymatic degradation of peptides refers to the breakdown of peptide chains by enzymes (proteases) that cleave the peptide bonds linking amino acids. Proteases are the primary agents responsible for peptide degradation, hydrolyzing amide bonds in the peptide backbone [1]. In biological systems, this process is ubiquitous – the human body encodes hundreds of proteolytic enzymes, and once a peptide is introduced, numerous proteases and peptidases can act on it. For example, after a peptide is absorbed into the bloodstream, it is exposed to a multitude of circulating proteases that rapidly begin to cleave it into smaller fragments [7]. This natural propensity for enzymatic breakdown means that many peptides have inherently short lifespans in vivo unless steps are taken to protect them from such degradation.
The Importance of Enzymatic Degradation Studies in Peptide Research
Enzymatic degradation studies are critically important in peptide research because peptide therapeutics often suffer from poor stability and low bioavailability due to rapid proteolysis. For instance, peptides delivered orally face harsh gastrointestinal (GI) conditions – stomach pepsin and intestinal enzymes can quickly digest them, making oral peptide delivery very challenging [9]. Likewise, even when delivered parenterally, circulating proteases and organ metabolism can inactivate peptides in minutes. This enzymatic degradation liability is a major stumbling block in peptide drug development; indeed, membrane impermeability and poor in vivo stability are intrinsic drawbacks of peptide therapeutics that limit their efficacy [3]. By conducting degradation studies, researchers can quantify how quickly a given peptide is destroyed by enzymes and thus gauge its peptide stability under physiological conditions.
Figure 1. Graphical depiction of peptide stability challenges, including enzymatic digestion in the gastrointestinal tract and metabolic degradation.
Enzymatic degradation studies provide essential data on peptide half-life and integrity over time, guiding the improvement of peptide candidates. Many native peptides exhibit extremely short circulation half-lives – on the order of only a few minutes – due to proteolysis [7]. For example, the peptide drug angiotensin II (Giapreza®) is metabolically unstable with an estimated half-life of only ~30 seconds in humans [7]. Such rapid clearance necessitates frequent dosing or continuous infusion, greatly limiting clinical utility. By evaluating stability early, scientists can identify these issues and implement strategies to extend peptide longevity. In fact, regulatory guidelines increasingly view proteolytic stability assessment as a fundamental part of peptide characterization, on par with evaluating potency, because ensuring sufficient stability is required for a peptide to maintain its bioactivity [2]. Overall, understanding how and how fast a peptide is degraded by enzymes informs medicinal chemists whether a lead peptide is viable as a drug or needs structural modification for better performance.
Common Enzymes Involved in Peptide Degradation
Peptide degradation in the body is mediated by a variety of proteolytic enzymes with specific cleavage preferences. In the GI tract, key enzymes include pepsin, trypsin, and chymotrypsin, among others. Pepsin (a stomach protease) cleaves peptide bonds preferentially at aromatic or hydrophobic residues (e.g. Phe, Trp, Tyr, Leu), whereas pancreatic trypsin hydrolyzes bonds on the carboxyl side of lysine or arginine, and chymotrypsin (a pancreatic serine protease) targets bulky hydrophobic residues like Phe, Tyr, Trp, Met, and Leu [6]. These enzymes readily attack susceptible peptide sequences during digestion, and the presence of their specific cleavage sites within a peptide makes it highly prone to GI degradation [6].
As a result, unmodified linear peptides are typically unstable in the digestive system. Beyond the GI tract, peptides encounter a broad spectrum of proteases throughout the body. The human protease repertoire is vast – there are over 600 proteases identified in humans [1] – and they are widely distributed in blood and tissues. In the bloodstream, for example, common peptidases include aminopeptidase N, carboxypeptidase N, dipeptidyl peptidase IV (DPP-4), as well as plasmin, furin, and neprilysin [7]. These enzymes rapidly attack circulating peptides from both ends or at specific internal sites, truncating the peptide chain. Essentially, without protective measures, a therapeutic peptide is subject to continuous assault by proteolytic enzymes – a fact that underscores why peptide degradation studies are so important for drug design.
Factors That Influence Enzymatic Stability
A peptide’s susceptibility to enzymatic degradation – its enzymatic stability – is determined by multiple intrinsic and extrinsic factors. Intrinsic factors include the peptide’s amino acid sequence, length, and conformation. Peptides containing sequences that are recognized cleavage sites for proteases will be broken down much faster. For example, if a peptide has many lysine or arginine residues, it becomes an easy target for trypsin; conversely, peptides that lack common cleavage sites or include residues that proteases struggle to cut (such as proline) tend to be more resistant to digestion [6]. In fact, incorporating certain residues can hinder enzyme action – studies have found that proline or acidic residues adjacent to cleavage motifs can improve resistance to enzymes like pepsin and pancreatin [6]. The length of the peptide also matters: large peptides or proteins (>3 kDa) often present more potential cut sites and tend to be hydrolyzed more extensively than very short peptides. Additionally, the secondary structure and overall conformation influence stability. A peptide that is cyclic or conformationally constrained may hide its scissile bonds from enzymes. Naturally occurring cyclic peptides (such as cyclotides) are notably resistant to proteolysis because their ring structure and disulfide bonds make them rigid and proteases cannot easily access their backbone [10].
Extrinsic factors relating to the environment also play a critical role in peptide stability. pH and enzyme environment can dictate which proteases are active and how aggressively they act. For example, the stomach’s low pH optimally activates pepsin, whereas pancreatic enzymes operate at neutral pH in the intestine. Likewise, the presence of enzyme cofactors or inhibitors, the temperature, and the incubation time all influence degradation. Stability analysis conditions used in experiments can dramatically affect the measured half-life of a peptide. Different research groups often use varying protocols – enzyme concentrations, peptide-to-enzyme ratios, incubation times, etc. – which can lead to variability in results [6]. This highlights that peptide stability is not a single fixed value, but rather a profile that can change with conditions.
Methods Used to Assess Enzymatic Degradation
Modern peptide research employs a range of methods to assess peptide stability against enzymatic breakdown. These include laboratory assays to mimic biological conditions and analytical techniques to detect peptide and fragment concentrations over time.
In Vitro Enzymatic Assays
In vitro enzymatic assays involve exposing the peptide to specific proteases or biological fluids under controlled conditions and monitoring its degradation. A simple approach is to incubate the peptide in a solution containing known enzymes (e.g., trypsin, chymotrypsin, or pepsin) or in plasma and measure how much of the intact peptide remains over time. Fluorogenic assays, for example, use labeled peptides that emit fluorescence when cleaved, allowing real-time monitoring [1]. Serum stability assays are another common format, incubating peptides in plasma at 37 °C to simulate physiological conditions; most peptides degrade within minutes unless stabilized [7]. These enzymatic assays yield valuable data such as half-life and cleavage patterns, guiding modifications to improve stability.
LC-MS and HPLC Analysis
Analytical techniques like high-performance liquid chromatography (HPLC) and liquid chromatography–mass spectrometry (LC-MS) are the gold standard for measuring peptide degradation and identifying fragments. Reversed-phase HPLC separates intact peptides from fragments, providing quantitative measures of degradation, while LC-MS identifies fragment masses and cleavage sites [1]. LC-MS data reveal degradation kinetics and map protease cleavage points, forming the foundation of degradation studies and quantitative stability analysis.
Enhancing Peptide Stability Through Design and Modification
Given the vulnerability of peptides to enzymatic attack, a major area of peptide science focuses on design strategies to enhance stability. A variety of molecular modification techniques have proven effective in making peptides more resistant to proteases. Broadly, these strategies aim to eliminate or shield the cleavage sites that enzymes recognize. For example, medicinal chemists often substitute natural L-amino acids in the peptide with D-amino acids (the mirror-image enantiomers). Proteases evolved to recognize the L-form of amino acids, so peptides containing D-amino acids at key positions become poor substrates for those enzymes and can evade degradation. Replacing even a few L-amino acids with D-amino acids in a peptide has been shown to dramatically improve its proteolytic stability and extend its half-life.
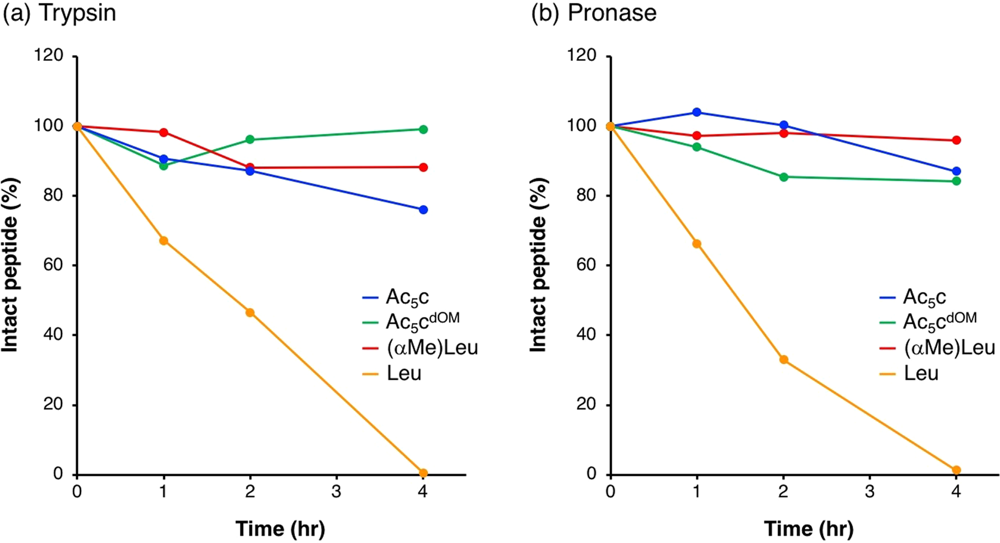
Figure 2. Comparative trypsin degradation profiles of peptide variants over time.
Another powerful strategy is cyclization of the peptide. By linking the N-terminus and C-terminus together (head-to-tail cyclization) or by introducing internal bridges (such as disulfide bonds or chemical staples), the peptide’s backbone is constrained in a loop. This conformational restriction can prevent proteases from binding and cutting the peptide as easily. Indeed, modifying peptides to form stable cyclic structures or “stapled” helices has yielded analogues with much greater resistance to enzymatic breakdown than their linear counterparts. Additional design approaches include N-terminal acetylation and C-terminal amide capping (to block exopeptidases from nibbling at the ends), incorporation of non-natural or unusual amino acids (e.g. N-methylated residues, β-amino acids, peptidomimetics) that proteases do not recognize, and conjugation of bulky groups like polyethylene glycol (PEGylation), which sterically hinders enzyme access and also reduces renal clearance.
These chemical modifications have proven successful in enhancing peptide stability and are often employed in combination. For instance, a peptide may be PEGylated and cyclized or contain both D-amino acid substitutions and a hydrocarbon staple, to maximize its half-life. The impact of such modifications is evident in the peptide therapeutics that have reached the market. A survey of approved peptide drugs found that the vast majority – 20 out of 25 analyzed – contained unnatural amino acids or other stability-enhancing features, underscoring how crucial these tweaks are for a peptide’s pharmacological success. One classic example is the peptide drug liraglutide, an analogue of GLP-1: it is modified with a fatty acid chain and other substitutions, which protect it from DPP-4 enzymatic degradation and give it a far longer half-life than native GLP-1. Similarly, octreotide (an analogue of somatostatin) is a cyclic peptide with D-amino acids; its engineered structure makes it stable enough to be a practical drug, whereas natural somatostatin is degraded within minutes. In research settings, scientists have demonstrated that even highly protease-sensitive peptides can be made stable. For example, converting a linear peptide into a cyclic one or “stapling” an α-helix can render it completely resistant to trypsin and other proteases for hours
Figure 2 shows an illustrative result: a linear peptide (orange line) is fully digested by trypsin within 4 hours, whereas analogues with constrained or modified structures (blue, green, red lines) remain largely intact over the same period【45†】. Such outcomes confirm that rational design can substantially boost peptide stability. Of course, these modifications must be balanced with maintaining the peptide’s biological activity and target binding, but many modern peptide drugs exemplify that it is possible to achieve both. Enzymatic degradation studies guide this process, as they allow researchers to test modified peptides and verify that the changes successfully imparted the desired resistance to proteases.
Enhancing Peptide Stability Through Design and Modification
Because peptides are vulnerable to enzymatic attack, structural modification is key to improving peptide stability. Approaches include substituting natural L-amino acids with D-amino acids, which proteases cannot recognize [4], cyclization to restrict backbone flexibility [10], and N-/C-terminal modifications like acetylation or amidation to protect against exopeptidases [4]. Other strategies, such as incorporating non-natural amino acids, PEGylation, or hydrocarbon stapling, further increase resistance to enzymatic degradation [8]. These modifications have led to dramatic improvements in peptide half-life and therapeutic viability. For example, liraglutide and octreotide use such chemical enhancements to resist enzymatic degradation and maintain biological activity [8]. Modern peptide drugs often integrate multiple modifications, as shown by the fact that most approved peptides contain unnatural amino acids or stability-enhancing features [5].
Applications of Enzymatic Degradation Studies in Drug Development
Enzymatic degradation studies play a multifaceted role in drug development research for peptide-based therapeutics. First and foremost, they serve as a screening tool in the early stages of development. When chemists design or discover a new bioactive peptide, one of the key questions is: Will this peptide remain intact long enough in the body to exert its effect? By subjecting candidate peptides to stability assays (in serum, GI fluids, etc.), researchers can triage which candidates are too unstable to be viable drugs. If a peptide is rapidly degraded, the studies will pinpoint the vulnerable sites (e.g. a specific bond consistently cleaved) and inform subsequent analog design. This iterative process – design → degradation test → redesign – is central to optimizing peptide leads. For example, if enzymatic assays reveal that a peptide is cleaved between two particular amino acids, medicinal chemists might modify that bond (e.g. use a non-cleavable isostere or D-amino acid) in the next iteration. In the case of oral peptide drugs, degradation studies are especially critical: the inherent enzymatic instability in the digestive tract has long been a major bottleneck, so extensive tests (including simulated gastric/intestinal digestion and in vitro gut models) are done to evaluate and improve oral peptide formulations. These studies might lead to protective strategies like enteric coatings, co-formulation with enzyme inhibitors, or use of nanoparticle delivery systems to shield the peptide through the GI transit.
Another important application of degradation studies is in establishing dosage forms and administration routes for peptide drugs. Stability analysis data, combined with pharmacokinetic modeling, help determine whether a peptide should be delivered via injection, infusion pump, or can be formulated as an oral or nasal delivery (depending on how quickly it is degraded in different compartments). For instance, if a peptide has a short half-life in blood, developers might consider a sustained-release injectable depot or modify the peptide (e.g. PEGylation) to prolong its circulation time. In clinical development, enzymatic degradation studies also inform toxicity and efficacy considerations – a peptide that degrades into inactive fragments might have fewer off-target effects, whereas one that degrades into bioactive fragments might pose risks or benefits that need evaluation. Moreover, regulatory agencies often expect stability data in the submission dossier for a peptide therapeutic, demonstrating that the sponsor has characterized the product’s metabolic stability and, if necessary, implemented measures to address rapid degradation. In summary, enzymatic degradation studies are indispensable throughout the pipeline of peptide drug development: from lead optimization (enhancing stability) to formulation (ensuring the peptide can reach its site of action) and ultimately to defining how the drug is used in patients (dose and route). By understanding and controlling peptide degradation, researchers can improve peptide performance, turning fragile peptide molecules into effective and reliable medicines.
Frequently asked questions (FAQs) about Peptide Isolation
What is enzymatic degradation and how does it affect peptides?
- Enzymatic degradation refers to the breakdown of peptide bonds by proteolytic enzymes such as trypsin, chymotrypsin, or peptidases. These enzymes recognize specific amino acid sequences and cleave the peptide chain, reducing structural integrity and biological activity. For therapeutic or research peptides, enzymatic degradation can significantly shorten their half-life and limit their effectiveness in biological systems [1].
Why are enzymatic degradation studies critical for peptide research?
- These studies are essential for determining a peptide’s stability, metabolic fate, and suitability for use in vivo. By understanding how quickly and through which pathways a peptide is degraded, researchers can optimize formulations, improve delivery systems, and design analogues with enhanced resistance to enzymatic cleavage. Such data are crucial for preclinical testing and regulatory approval [2].
What factors influence peptide stability and enzyme resistance?
- Peptide stability depends on multiple factors, including sequence composition, secondary structure, terminal modifications, and environmental conditions such as pH, temperature, and ionic strength. Certain amino acid residues make peptides more susceptible to enzymatic attack, while chemical modifications—like cyclization, N-methylation, or incorporation of D-amino acids—can improve resistance to degradation [3].
How do scientists assess and measure enzymatic degradation?
- Researchers use analytical techniques such as high-performance liquid chromatography (HPLC), mass spectrometry (MS), and electrophoretic assays to monitor peptide integrity over time. Enzyme incubation assays help quantify degradation kinetics and identify cleavage sites. Circular dichroism and fluorescence spectroscopy may also be used to evaluate structural changes during digestion [4].
How can peptide design reduce susceptibility to enzymatic breakdown?
- Rational peptide design can greatly improve enzyme resistance. Strategies include cyclizing the peptide backbone to restrict flexibility, substituting natural amino acids with non-canonical or D-amino acids, and capping terminal ends to prevent exopeptidase activity. Modern computational modeling allows scientists to predict vulnerable regions and optimize structural modifications before synthesis, enhancing peptide stability without sacrificing biological function [5].
References
-
Tan X, et al. Enzyme-catalyzed cleavage reactions as primary pathways for peptide degradation. Brief Bioinform. 2024;25(4):bbae350.
-
Pan D, et al. Poor proteolytic stability and rapid metabolism limit peptide efficacy. Curr Res Food Sci. 2023;6:2535–2547.
-
Scudamore CL, et al. Intrinsic drawbacks of peptides include membrane impermeability and poor in vivo stability. Signal Transduct Target Ther. 2022;7(1):254.
-
Oba M, et al. Synthetic modifications (D-amino acids, stapling) confer resistance to proteolytic degradation. Sci Rep. 2019;9(1):1349.
-
Chia JH. Unnatural amino acid substitution extends peptide half-life; most approved peptide drugs contain stability-enhancing modifications. Pharmaceutics. 2021;13(10):1662.
-
Udenigwe CC, et al. Proline and acidic residues improve peptide resistance to GI enzymes. Food Chem. 2015;171:15–23.
-
Bottger R, et al. Serum stability assays show most peptides are rapidly degraded in blood unless structurally modified. Eur J Pharm Biopharm. 2017;115:47–56.
-
Al Musaimi O, et al. Strategies for improving peptide stability: D-amino acids, stapling, and cyclization. Pharmaceutics. 2022;14(1):120.
-
Zhang P, et al. Peptide susceptibility to enzymatic degradation in the GI tract and its role in oral drug development. Acta Pharm Sin B. 2021;11(8):2416–2435.
-
Muttenthaler M, et al. Cyclotides and cyclic peptides as stable scaffolds against proteolysis. Nat Rev Drug Discov. 2021;20(8):522–540.