
LPS-Binding Proteins and Their Role in Endotoxin Detection
Author: Dr. Numan S. Date: January 15, 2026
What Is Endotoxin (LPS) and Why Does Detection Matter?
Endotoxin refers to lipopolysaccharide (LPS), a structural component of the outer membrane of Gram-negative bacteria that elicits potent immune responses in mammals [1,2]. Chemically, LPS is composed of a lipid A domain, which is responsible for its toxic activity, a core oligosaccharide, and in many species an O-antigen polysaccharide chain [3,4].
When LPS enters the bloodstream—such as through contaminated injectable or parenteral products—it can trigger fever, systemic inflammation, hypotension, and in severe cases septic shock [1,3]. Because endotoxin remains biologically active even after bacterial death or sterilization, its detection is a critical safety requirement in pharmaceutical and medical research workflows. Regulatory frameworks, including USP <85>, require routine endotoxin testing of sterile products prior to release [1].
What Are LPS-Binding Proteins?
LPS-binding proteins are biological or assay-associated proteins that recognize and bind lipopolysaccharide, typically through interactions with the lipid A region [2,3]. In mammalian systems, the best-characterized example is lipopolysaccharide-binding protein (LBP), a ~60 kDa acute-phase glycoprotein synthesized primarily by the liver [2].
LBP binds aggregated LPS and extracts monomeric endotoxin molecules, facilitating their transfer to downstream receptors involved in immune signaling. Additional host proteins involved in LPS recognition include CD14, MD-2, and Toll-like receptor 4 (TLR4), which together form the core endotoxin-sensing machinery of the innate immune system [3]. Antimicrobial proteins such as bactericidal/permeability-increasing protein (BPI) and lactoferrin also bind LPS, often neutralizing its biological activity [2].
Overview of Common Endotoxin Detection Methods
Several methods are used to detect endotoxins, and each has implications for LPS-binding interactions:
-
Rabbit Pyrogen Test (RPT): An older method (historically the first FDA-approved endotoxin test) in which a sample is injected into rabbits, and the animals’ body temperature is monitored. A fever indicates pyrogens (like endotoxin) are present. This in vivo test inherently detects biologically active LPS but is slow and uses live animals. It has largely been supplanted by in vitro assays.
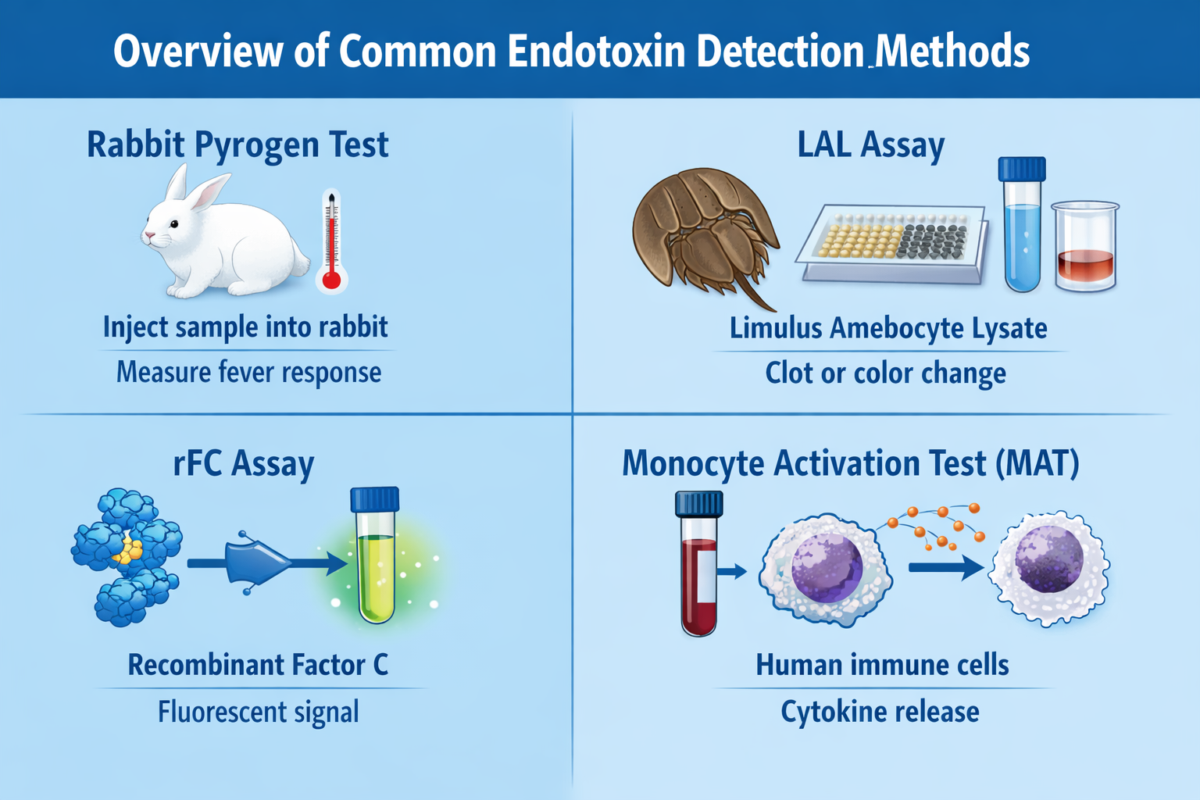
Figure 1: Overview of Common Endotoxin Detection Methods
- Limulus Amebocyte Lysate (LAL) Assay: The LAL assay is the long-standing gold standard for endotoxin detection. It uses lysate from horseshoe crab blood cells (amebocytes) which contains a cascade of LPS-sensitive proteins. Factor C in the lysate binds any endotoxin in the sample, triggering a cascade (through Factor B and a proclotting enzyme) that leads to either gel clot formation or a color change (depending on the assay format). The LAL assay is extremely sensitive (detecting endotoxin in the low EU/mL range). However, it can be affected by non-endotoxin substances: for example, (1→3)-β-D-glucans from fungi activate Factor G in LAL, causing false positives. Despite minor limitations, LAL remains widely used in pharma due to its sensitivity and extensive validation history.
-
Recombinant Factor C (rFC) Assay: The rFC assay is a modern alternative that addresses some LAL drawbacks. It uses a single recombinant LPS-binding protein (Factor C) and a synthetic fluorogenic substrate. When Factor C binds endotoxin, it directly cleaves the substrate to produce a fluorescent signal. By design, rFC assays exclude the other crab blood components (like Factor G), making them specific to LPS and avoiding false activation by glucans. They also have ethical and sustainability advantages, as they do not require bleeding horseshoe crabs. Studies show rFC results correlate well with LAL and even have improved endotoxin recovery in some cases. In fact, regulatory authorities (e.g., European Pharmacopeia) have begun to accept rFC-based methods as equivalents to LAL for endotoxin testing.
-
Monocyte Activation Test (MAT): This is an ex vivo assay that uses human blood cells or a cell line to detect pyrogens. The principle is that human immune cells will respond to endotoxin (and other pyrogens) by releasing cytokines. The released cytokines (like IL-6) are then measured (often by ELISA). MAT has the advantage of detecting a broad range of pyrogens (not just LPS) and mimicking the human fever reaction. It’s used especially when a holistic pyrogen test is needed or when LAL/rFC might not be sufficient (for example, to detect non-endotoxin pyrogens). However, MAT is more complex to perform and not as high-throughput as LAL/rFC.
-
Other methods: Emerging or specialized methods include ELISA kits with LPS-specific antibodies and PCR-based detection of bacterial DNA (as an indirect endotoxin proxy). These are less common for routine endotoxin testing but may appear in research contexts.
LPS-Binding Effects That Can Impact Assay Results
While endotoxin detection assays are powerful, various factors related to LPS binding can introduce assay interference. One major issue is endotoxin masking – when LPS in a sample is bound or sequestered by other components, making it “invisible” to the detection reagent. For example, protein solutions or blood products may contain LPS-binding proteins (like antibodies, albumin, or LBP itself) that bind endotoxin and prevent the LAL or rFC reagents from accessing it. This leads to falsely low readings. Certain formulations in biopharmaceuticals (e.g. buffers with polysorbate detergent and citrate) can cause LPS to form complexes with proteins, resulting in low endotoxin recovery (LER) – a phenomenon where endotoxin that is deliberately spiked into a product cannot be recovered by the LAL test. LER is a known challenge, as it may cause a contaminated sample to falsely pass as “endotoxin-free” if the endotoxin is masked.
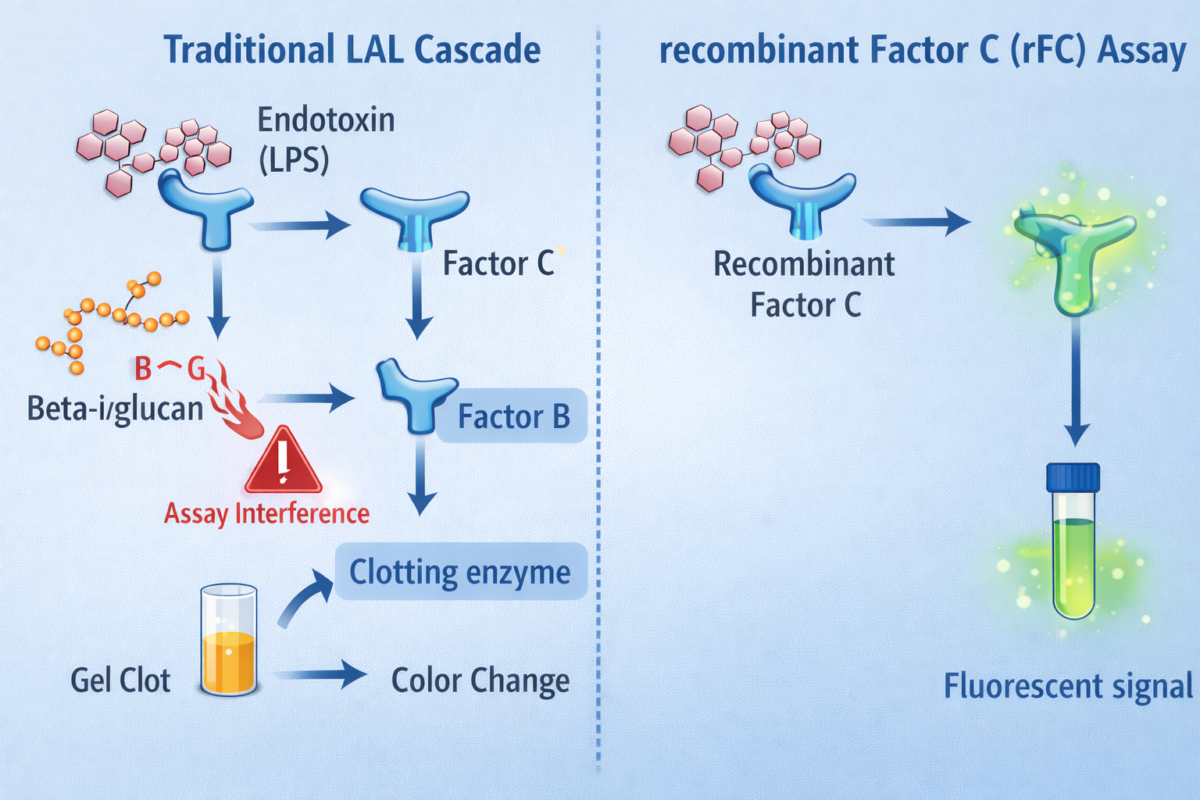
Figure 2: Diagram comparing the LAL cascade and rFC assay, highlighting how LPS and beta-glucan influence detection and assay interference.
Besides masking (inhibition), the opposite problem is assay enhancement – substances that make an assay respond more strongly. An example is the presence of β-glucans from fungal cell walls triggering the Factor G pathway in LAL, which yields a clot even if no endotoxin is present. This cross-reactivity is essentially a false positive. Similarly, extreme pH or chelating agents in a sample can alter LAL results: EDTA, for instance, chelates divalent cations; this can both reduce LPS aggregation (making LPS more reactive) and inhibit the enzymes in the LAL cascade, causing unpredictable effects. Proteolytic enzymes in samples might degrade LAL proteins (inhibiting the assay), whereas other enzymes (e.g. trypsin) might prematurely activate the cascade and give false positives.
Ultimately, any factor that changes LPS’s availability or the reaction of the detection proteins can impact results. That’s why regulatory guidelines insist on thorough method suitability testing (spike recovery experiments) to check for inhibition or enhancement in each new sample type.
Best Practices for More Reliable Endotoxin Detection
For researchers and QC teams, awareness of LPS-binding interactions leads to several best practices to ensure reliable endotoxin detection:
-
Perform Spike Recovery Tests: Always run inhibition/enhancement controls by spiking a known amount of endotoxin into your sample matrix. This challenge test (part of method validation) will reveal if your sample’s components bind LPS or interfere with the assay. Acceptable recovery is typically 50–200% of the spiked value; anything outside indicates interference requiring attention.
-
Dilute or Treat Samples to Mitigate Interference: Dilution is a simple and effective way to reduce interference. By diluting a sample, you decrease the concentration of LPS-binding proteins or other inhibitors, making any endotoxin more detectable (as long as you stay within the assay’s sensitivity range). If dilution alone isn’t enough, other treatments can help. For protein-rich samples, heat treatment (e.g. 70–80°C for a short time) can denature proteins like LBP or enzymes without destroying LPS, thus “unmasking” bound endotoxin. Similarly, adjusting the sample pH into the optimal range for the LAL/rFC enzymes, or adding magnesium/calcium if chelators are present, will improve assay performance.
-
Consider Alternative Assays: If the standard LAL assay is yielding inconsistent results due to interference, consider using the rFC assay or MAT. The rFC assay’s specificity for endotoxin (no Factor G pathway) can bypass certain false positives and may handle some matrices better. The monocyte activation test can detect endotoxin even if it’s masked in a formulation, because human cells may still respond to LPS that enzymatic assays can’t detect. In some cases, switching to an endotoxin-specific ELISA or a kinetic turbidimetric method might also help, depending on the interference profile.
-
Remove or Neutralize Endotoxin Upfront (when feasible): If your goal is to ensure a sample is endotoxin-free (rather than measuring how much is present), you can use agents like polymyxin B resin to bind and remove LPS prior to final testing. Polymyxin B is an antibiotic that tightly binds lipid A. Be cautious, though: for an assay meant to quantify endotoxin, you shouldn’t remove it completely. However, polymyxin B can be employed in sample preparation to reduce high endotoxin loads or to demonstrate that an observed effect is due to endotoxin (by comparing results with and without polymyxin treatment).
By integrating these practices – validating assays with controls, mitigating interference factors, and choosing the right detection method – researchers can greatly improve their confidence in endotoxin detection. Awareness of how LPS-binding proteins and other components interact with endotoxin helps avoid false negatives or positives, ensuring that contamination is accurately detected and patient safety is protected.
Frequently asked questions (FAQs) about LPS-Binding Proteins and Their Role in Endotoxin Detection
What are LPS-binding proteins and what do they do?
- LPS-binding proteins are molecules that specifically recognize and bind lipopolysaccharide (LPS), the endotoxin component of Gram-negative bacterial outer membranes. In biological systems, proteins such as lipopolysaccharide-binding protein (LBP), CD14, and components of the MD-2/TLR4 complex bind LPS and facilitate immune recognition and signaling. In analytical contexts, LPS-binding interactions can either enhance or inhibit endotoxin detection depending on whether the binding exposes or masks the lipid A region responsible for endotoxin activity.
How do LPS-binding interactions influence endotoxin detection?
- LPS-binding interactions influence endotoxin detection by altering the availability of lipid A, the bioactive portion of LPS that most assays detect. When LPS is bound to proteins, lipoproteins, or surfactants, its reactive sites may be partially shielded, reducing assay sensitivity. Conversely, certain binding proteins can present LPS in a way that enhances detection by facilitating interaction with assay reagents, such as Factor C in limulus-based assays. These interactions are a key reason why endotoxin results can vary between assay formats or sample matrices.
Why can endotoxin be difficult to detect in complex samples?
- Endotoxin can be difficult to detect in complex samples because LPS readily associates with proteins, lipids, polymers, and particulate matter. These interactions can mask endotoxin activity, inhibit assay enzymes, or interfere with reagent binding. Additionally, sample components such as buffers, salts, or solvents may suppress or enhance assay signals, leading to false negatives or inconsistent results. Matrix effects are particularly relevant in peptide, protein, and biologics research where endotoxin may be present but not freely accessible.
What are the differences between LAL and recombinant assays?
- Limulus amebocyte lysate (LAL) assays use enzymes derived from horseshoe crab blood to detect endotoxin through activation of Factor C, while recombinant assays use genetically engineered recombinant Factor C (rFC). LAL assays can be susceptible to interference from β-glucans and other substances unless properly controlled, whereas rFC assays are more specific to endotoxin and eliminate reliance on animal-derived reagents. Recombinant assays also tend to offer improved consistency and reduced variability in complex research samples.
What best practices reduce false negatives or misleading results?
- Best practices for reducing false negatives or misleading endotoxin results include validating assays for each sample matrix, performing dilution or spike-recovery studies, and using endotoxin-free reagents and labware. Including appropriate positive controls helps confirm assay performance in the presence of potential inhibitors. Selecting an assay format suited to the sample type and minimizing protein or surfactant interference during sample preparation further improves detection accuracy. Consistent handling and proper storage of samples also reduce variability in endotoxin measurements.
References
-
Sandle T. Bacterial Endotoxin Test using LAL methodology: overcoming interfering factors. European Pharmaceutical Review. 2021;Issue 4:104-113.
-
Piehler M, et al. Comparison of LAL and rFC Assays—Participation in a Proficiency Test Program 2014–2019. Microorganisms. 2020;8(3):418.
-
Frontiers in Microbiology (Maeshima & Fernandez, 2013). Recognition of lipid A variants by the TLR4–MD-2 receptor complex. Front Cell Infect Microbiol. 2013;3:3.
-
Wako Chemicals USA. The biochemical structure of LPS and how it acts in nature vs. the lab. (Blog). 2025.
-
BMG Labtech. The LAL assay: a living fossil exploited to detect bacterial contamination. (Blog). 2022.