
Peptide Fragmentation Patterns in Mass Spectrometry
Author: Dr. Numan S. Date: November 6, 2025
What Is Peptide Fragmentation?
Peptide fragmentation in mass spectrometry is the process of breaking a peptide ion into smaller fragment ions within the mass spectrometer [1]. In a tandem MS experiment (MS/MS), selected peptide precursor ions are induced to fragment (usually by collision with an inert gas), and the resulting product ions are then analyzed for their mass-to-charge ratios. This produces a fragmentation spectrum – a unique set of peaks corresponding to the masses of the fragments – which serves as a fingerprint of the peptide’s amino acid sequence [1]. Fragmentation is crucial for peptide identification in proteomics: without fragmenting the peptide, one would know only its intact mass, which often is not enough to determine the exact sequence among many possibilities [1]. By examining the pattern of fragment ions, scientists can deduce the peptide’s sequence or confirm its identity by matching the fragmentation spectrum to databases in proteomics analysis.
How Peptide Fragmentation Works in Mass Spectrometry
Peptide fragmentation typically occurs in the collision cell of a tandem mass spectrometer. In a common workflow, peptides are first ionized (e.g. by ESI or MALDI) and a precursor peptide ion of interest is selected in the first stage (MS1). This ion is then injected into a collision region and bombarded with neutral gas molecules (like nitrogen or argon). The collisions supply energy to the ion, causing it to break apart into smaller ions (fragments) at various bonds along the peptide backbone [2]. Each fragmentation event can occur at different peptide bonds somewhat randomly (though some bonds break more readily than others), so a single peptide often yields a series of fragment ions of different lengths [2]. For instance, breaking one specific amide bond produces two complementary fragments – one carrying the N-terminal part and another carrying the C-terminal part of the peptide. Collectively, multiple fragmentation events generate a set of ions (fragments differing by one amino acid or several amino acids) that span the peptide’s length. These fragments are then separated by their m/z in the second mass analyzer (MS2) and detected as a fragmentation spectrum.
Each peak in a fragmentation spectrum corresponds to a fragment ion with a specific m/z. Importantly, the pattern of peaks (fragmentation pattern) is characteristic of the peptide’s sequence. Because each breakage yields a pair of complementary ions (one from the N-terminus side and one from the C-terminus side of the cleaved bond), an ideal fragmentation will produce a series of complementary ions that, together, cover the entire peptide sequence [4]. In practice, not all possible bonds break or produce observable ions, but typically a seriesof prominent fragment ions can be detected. By analyzing the mass differences between these fragment ions, the sequence of amino acids can be inferred. This forms the basis of peptide analysis by tandem mass spectrometry in proteomics: the fragmentation spectra are matched to peptide sequences to achieve peptide identification.
Types of Fragment Ions in Peptide Spectra
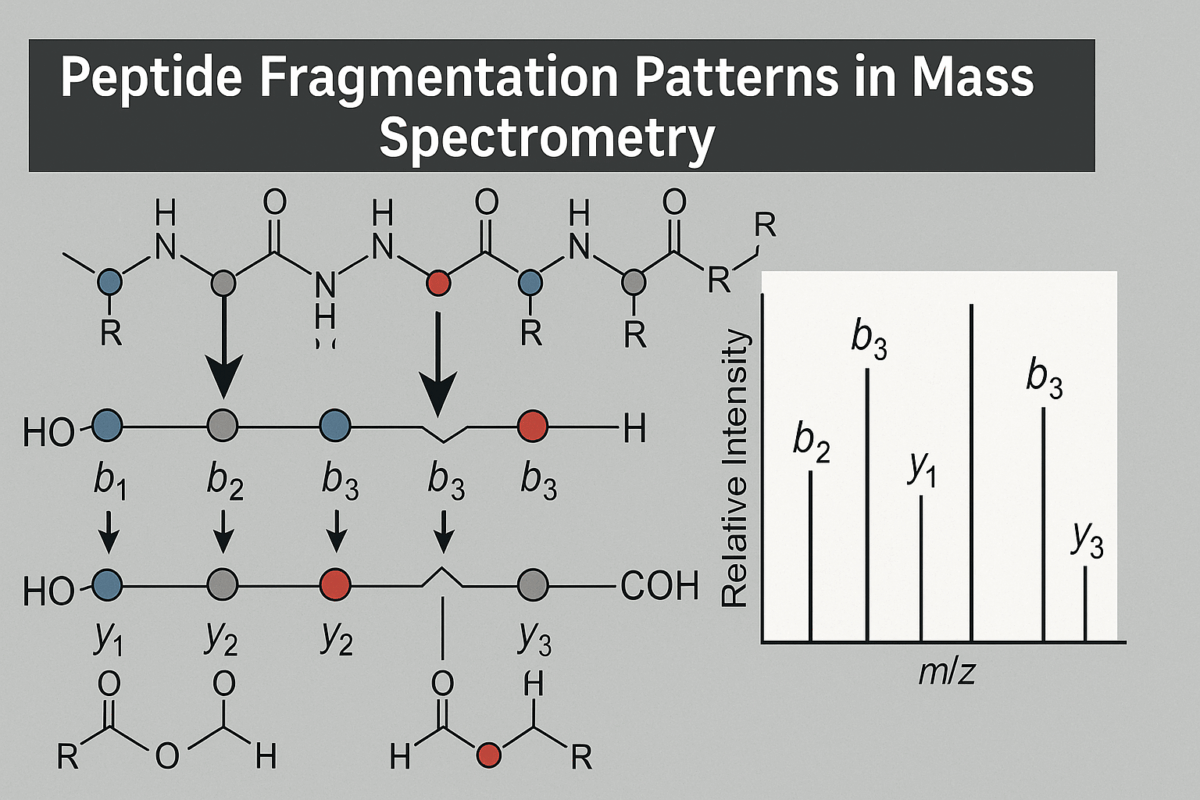
When a peptide ion fragments, the nomenclature of the resulting ions was defined by Biemann and others to indicate which part of the peptide the ion contains[6]. The six fundamental ion types from backbone cleavage are labeled a, b, c, x, y, z. If the charge is retained on the N-terminal side of the broken bond, the ions are in the a, b, or c series (with a being a fragment that has lost a CO group relative to b, and c having an additional NH group) [6]. If the charge remains on the C-terminal side, the ions belong to the x, y, or z series (with x and z being analogs on the C-terminal side) [6]. In most mass spectrometry of peptides, the b-ions and y-ions are by far the most commonly observed (especially under CID/HCD conditions). For example, a b ion means the fragment ion containing the first 5 amino acids from the N-terminus (with a proton making it positively charged), whereas a y ion contains the last 5 amino acids from the C-terminus (plus a proton). These two would be complementary fragments produced by cleavage at the peptide bond that divides the peptide into those two parts.
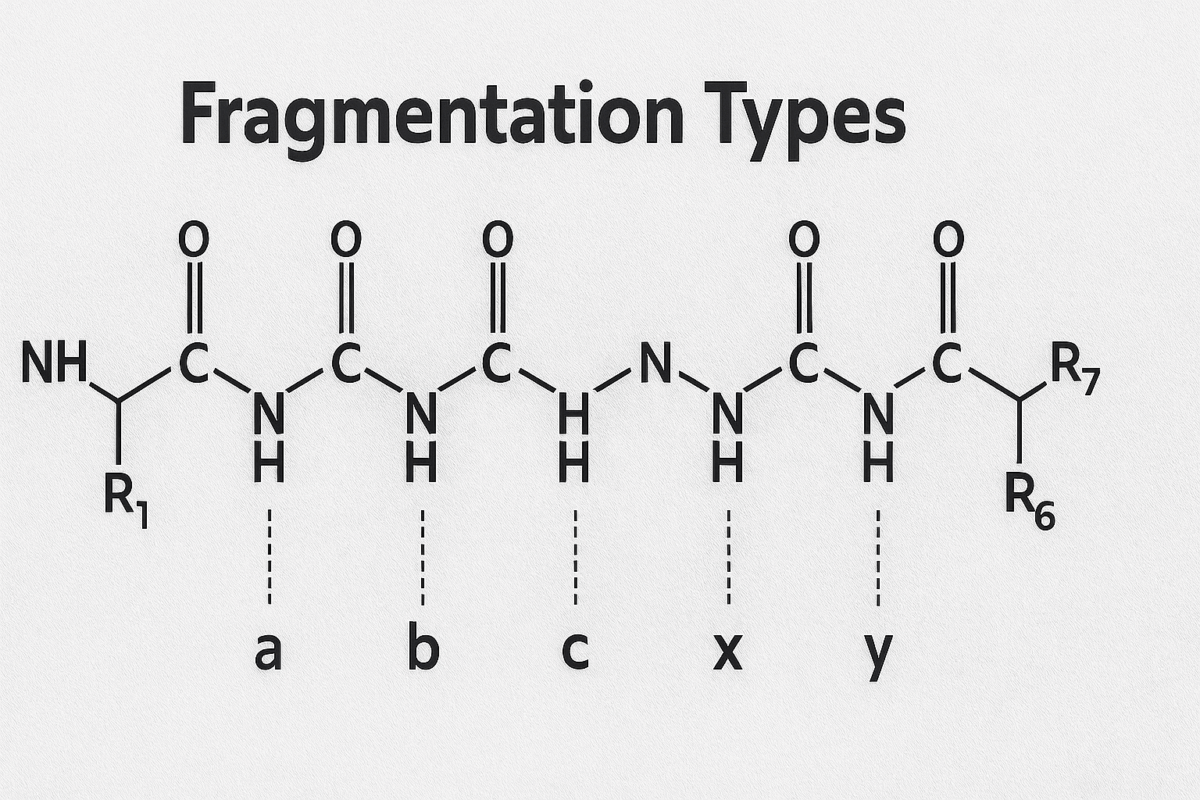
Figure 1: Visual representation of fragmentation types
In a typical fragmentation spectrum, one expects to see a ladder of b-ions rising in m/z as the fragment length increases, and a complementary ladder of y-ions (whose masses, when added to the corresponding b-ion, roughly equal the precursor mass plus an extra water molecule). For example, if a peptide has sequence ABCDE (5 amino acids), fragmentation could produce b through b ions (A, AB, ABC, ABCD) and y through y ions (E, DE, CDE, BCDE). The “b-ions and y-ions” are complementary: for instance, the mass of b plus the mass of y will equal the mass of the intact peptide (plus one water, 18 Da, because a y-ion by definition includes the elements of an extra HO) [6]. Recognizing these complementary pairs is very useful in spectrum interpretation, as it confirms the sequence coverage. The dominance of b/y ions is a fortunate feature of collision-based peptide fragmentation, as it provides clear sequence information in most cases. Meanwhile, in ETD or ECD spectra, one would mainly see c-ions and z-ions instead (which serve a similar purpose for sequence analysis, but with the roles of N- and C-termini reversed in terms of which part carries the charge).
Interpreting Fragmentation Spectra
Interpreting a peptide’s fragmentation spectrum means determining which peaks correspond to which fragment ions, and deducing the peptide’s sequence (or confirming a proposed sequence) from this information. A common approach is to look for a series of peaks that differ by the masses of common amino acid residues – this indicates a ladder of b-ions or y-ions. For example, if you see peaks that are spaced ~ 129 Da apart (which is the mass of glutamic acid minus H2_2O, 129), it suggests that glutamic acid might be one of the residues between those fragmentation points. In practice, automated algorithms compare the observed fragmentation spectra to theoretical spectra of candidate peptides from a database, scoring matches based on the presence of expected b/y ions. A spectrum with a high number of matching b- and y-ions to a particular sequence is strong evidence that the peptide identification is correct. Conversely, de novo sequencing algorithms attempt to read the sequence directly by interpreting gaps between fragment masses.
A well-interpreted fragmentation spectrum will show complementary b-ion and y-ion pairs. The highest m/z y-ion often corresponds to the loss of a single amino acid from the peptide (y1_1 ion, usually a basic residue if the peptide was tryptic), and the lowest m/z b-ion (b1_1) corresponds to the N-terminal amino acid (often not observed if it’s very small). The sum of a b-ion’s m/z and its complementary y-ion’s m/z (plus 1 Dalton for the hydrogen difference) should equal the precursor’s m/z plus the mass of water [6]. Checking these relations helps ensure the observed ions are correctly assigned. For instance, if a b5_5 ion and a y4_4 ion are both present in the spectrum of a 9-residue peptide, the masses of those ions should add up to the precursor mass (within a small error). This complementary pair property is a hallmark of high-quality fragmentation data and is used by algorithms to confidently label peaks as b or y ions [4].
Interpreters also look for diagnostic ions. Immonium ions (at very low m/z, e.g. 110 for histidine, 136 for tyrosine) indicate the presence of certain residuesmatrixscience.com. If specific neutral loss peaks are present (like a strong peak at −98 Da from a y-ion, indicating loss of H3_3PO4_4, common for phosphoserine/threonine), that can hint at a phosphorylation modification. In spectra obtained by ETD, the presence of a z˙^{dot{}} ion series (often noted by an odd-numbered m/z due to the radical) alongside c-ions would be interpreted differently but similarly yields sequence information.
Manually piecing together a peptide sequence from a spectrum involves adding up mass differences: e.g., a gap of 113 Da between b3_3 and b4_4 implies the 4th residue is 113 Da (likely leucine or isoleucine, which are isobaric). Modern software will do this systematically, but understanding the process is important for validation. Peptide fragmentation spectra can sometimes be complex (especially if multiple charge states or unexpected ions appear), but under typical conditions, the dominant peaks correspond to b/y ions that allow the sequence to be read in order. Ideally, one can find a continuous series of either b-ions from the N-terminus or y-ions from the C-terminus (or both) that cover the full length of the peptide. Many spectra, in practice, show a mix – for example, b-ions dominate lower m/z and y-ions dominate higher m/z. The goal is to interpret enough of them to either uniquely identify the peptide or confirm that a proposed peptide matches the observed pattern.
Notably, advanced interpretation methods including machine learning have been applied to classify and predict fragment ions. For instance, algorithms can be trained to distinguish b-ion peaks from y-ion peaks in a spectrum, improving de novo sequencing accuracy [4]. As fragmentation is a reproducible physical process, it’s possible to predict likely fragmentation patterns for a given sequence; this is used in in silico spectral libraries and scoring functions to improve peptide identification.
Types of Fragment Ions in Peptide Spectra
When a peptide ion fragments, the nomenclature of the resulting ions was defined by Biemann and others to indicate which part of the peptide the ion contains. The six fundamental ion types from backbone cleavage are labeled a, b, c, x, y, z. If the charge is retained on the N-terminal side of the broken bond, the ions are in the a, b, or c series (with a being a fragment that has lost a CO group relative to b, and c having an additional NH2_2 group). If the charge remains on the C-terminal side, the ions belong to the x, y, or z series (with x and z being analogs on the C-terminal side).
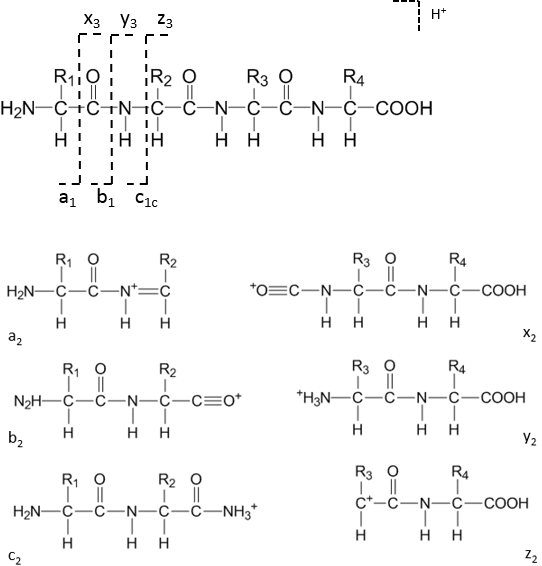
Figure 2.Eptide backbone cleavage positions and fragment ion types
In most mass spectrometry of peptides, the b-ions and y-ions are by far the most commonly observed (especially under CID/HCD conditions). For example, a b5_5 ion means the fragment ion containing the first 5 amino acids from the N-terminus (with a proton making it positively charged), whereas a y5_5 ion contains the last 5 amino acids from the C-terminus (plus a proton). These two would be complementary fragments produced by cleavage at the peptide bond that divides the peptide into those two parts.In summary of the nomenclature: starting from the N-terminus, a peptide of sequence N-[1][2][3]…[(n)]-Ctext{N-}[1][2][3]…[(n)]text{-C} can fragment to yield: ai_i, bi_i, or ci_i ions (for i=1 to n−1) which contain the first i amino acids (N-terminal side); and xj_j, yj_j, or zj_j ions (for j=1 to n−1) which contain the last j amino acids (C-terminal side). The most common fragmentation in conventional proteomics generates b-ions (N-terminal fragments minus a hydrogen) and y-ions (C-terminal fragments plus a hydrogen and H2_2O). Minor ion types like a-ions (which are b-ions minus CO, typically a result of further fragmentation of b-ions) or z˙^{dot{}} and x-ions (from ECD/ETD or high-energy processes) may also appear, but in low-energy CID spectra a-ions are occasionally seen whereas x-ions are rare. Other specialized fragments include internal ions (resulting from multiple backbone breaks, yielding a middle portion of the sequence) and immonium ions (single-residue marker ions), but these are usually small peaks used for specific diagnostic purposes.
Factors That Influence Peptide Fragmentation Patterns
Several factors can influence the fragmentation pattern of a peptide in mass spectrometry, affecting which bonds break and how intense certain fragment ions are. One major factor is the peptide’s charge state. Peptides that carry more charges (higher protonation states) often fragment more easily (requiring less energy per bond to dissociate) and can exhibit different fragmentation pathways. For example, a doubly protonated peptide in CID might produce both b and y ions readily, whereas a singly protonated peptide might favor either N-terminal or C-terminal fragmentation predominantly, due to where the proton is located (the mobile proton model) [4]. Peptide length and mass also play a role: larger or heavier peptides have more bonds and typically require more energy (or multiple steps) to fragment completely. They may not yield a full series of fragments with a single fragmentation event. This is one reason why longer peptides or whole proteins are often fragmented by multiple methods or multiple stages (as in top-down proteomics or multi-step MS^n) to cover the entire sequence.
The amino acid sequence and composition strongly influence fragmentation. Certain residues create preferential or weak breakpoints. For instance, the presence of a proline residue tends to “direct” fragmentation to occur on its N-terminal side – peptides often show enhanced cleavage at the bond N-terminal to prolinematrixscience.com. Acidic residues (Asp, Glu) and imidic residues (Pro) can affect fragmentation by mechanisms like promoting specific charge locations or forming cyclic intermediates (e.g. the “proline effect” causes intense y-ions because the proline-containing fragment is stabilized)matrixscience.com. Basic residues (Lys, Arg, His) if located near the termini can sequester protons and alter the pattern (e.g. a peptide with a C-terminal Lys/Arg often yields a strong y1_1 ion corresponding to that single basic residue at the C-terminus). The distribution of mobile protons along the peptide chain (dictated by the presence of basic sites) is known to govern which bonds break – bonds adjacent to where a proton can attach become more susceptible to cleavage (according to the mobile proton model of fragmentation). Additionally, certain sequence motifs (like Asp-Pro) are known to undergo specific cleavages.
Applications of Peptide Fragmentation Analysis
Understanding and analyzing peptide fragmentation patterns is central to proteomics analysis. The primary application is peptide identification: in bottom-up proteomics, proteins are digested into peptides, and these peptides are identified by matching their MS/MS fragmentation spectra against peptide databases. The confidence in identification comes from how well the experimental fragmentation pattern matches the theoretical pattern of a candidate peptide. Database search engines (like SEQUEST, Mascot, etc.) and newer algorithms (like machine learning-based predictors) all rely on the principles of peptide fragmentation to score matches. A rich fragmentation spectrum with many b-ions and y-ions will yield a high score and a confident identification, whereas a poor or incomplete fragmentation pattern might lead to ambiguity.
Another key application is de novo sequencing of peptides, where no database match is assumed. Here, the fragmentation pattern itself must be decoded to yield the sequence. Successful de novo sequencing requires a near-complete series of fragment ions. Techniques that improve fragmentation coverage (like using HCD to get a and b ions at low mass and y ions at high mass, or combining CID and ETD results) make de novo identification more feasible. Specialized methods like pseudo-MS^3 (where a specific ion is fragmented further) can help resolve sequence ambiguities for de novo analysis.
Peptide fragmentation analysis is also crucial for localizing post-translational modifications (PTMs). For example, if a peptide is phosphorylated, the fragmentation spectrum can indicate which fragment ions still carry the modification. A series of y-ions might suddenly drop 98 Da in mass at a certain position, pinpointing the phosphate loss and hence the modified residue. ETD is particularly valuable for this, as it often preserves modifications on c and z ions, allowing clear localization. In proteomics, identifying not just the peptide sequence but also any modifications and their exact sites is often necessary, and fragmentation patterns provide that information (e.g., a neutral loss of 64 Da might indicate a loss of sulfate from a tyrosine, etc.).
Furthermore, fragmentation patterns are used in targeted proteomics approaches. In Selected Reaction Monitoring (SRM) or Parallel Reaction Monitoring, specific fragment ions of a peptide are monitored as signatures for the presence of a particular peptide (and by extension, the protein it came from). These fragment ions (often y-ions due to their reliability) are chosen based on predictable fragmentation patterns. The consistency of peptide fragmentation enables us to develop libraries of such transitions for quantitative analyses.
Lastly, fragmentation analysis is used in the characterization of novel peptides or peptide-based drugs. In pharmaceutical analysis, for instance, if a new peptide drug is being analyzed, tandem MS fragmentation can confirm its sequence and detect any unexpected modifications or degradation products by the pattern of fragments. It’s also used in peptidomics (the study of endogenous peptides in cells/tissues) to identify and quantify peptides present in biological samples.
Frequently asked questions (FAQs) about Peptide Fragmentation
How does peptide fragmentation occur in mass spectrometry?
- Peptide fragmentation occurs when peptide ions are energized and break along their backbone bonds, typically between the carbonyl carbon and amide nitrogen. This process generates smaller ion fragments that represent portions of the original peptide sequence, which can then be analyzed to deduce its structure and amino acid composition [1].
What are the key ion types produced during fragmentation?
- The most common fragment ions are b-ions and y-ions formed from cleavage at the amide bond. b-ions retain the N-terminal portion, while y-ions retain the C-terminal portion of the peptide. Other less frequent ion types, such as a-, c-, x-, and z-ions, can also appear depending on the fragmentation method used [2].
How can fragmentation spectra be used to identify peptides?
- Each peptide has a unique fragmentation pattern that serves as a “fingerprint.” By comparing the observed fragment masses against theoretical peptide databases, researchers can determine the peptide’s sequence and confirm protein identity in proteomic studies [3].
What factors affect fragmentation efficiency and pattern?
- Fragmentation depends on multiple factors, including peptide length, charge state, sequence composition, and the energy of collision or dissociation. Amino acids like proline and aspartic acid often promote specific cleavage sites, while peptides with higher charge states tend to fragment more efficiently [4].
How do CID and ETD fragmentation methods differ?
- Collision-Induced Dissociation (CID) fragments peptides through high-energy collisions with inert gas molecules, mainly producing b and y ions. Electron Transfer Dissociation (ETD), in contrast, uses electron transfer reactions to break the peptide backbone while preserving post-translational modifications, making it ideal for mapping modified or complex peptides [5].
References
- Lee A. Freedom to Fragment: Introduction. Thermo Fisher Scientific AnalyteGuru Blog. 2025 Jul 30. thermofisher.com
- OHSU Proteomics Core. Identifying Proteins: from Gel Bands to Useable Data (technical document). Oregon Health & Science University; 2019. ohsu.edu
- Matrix Science. Mascot Database Search: Peptide Fragmentation. Matrix Science Ltd. (Mascot documentation). Accessed 2025. matrixscience.com
- Li X. Classification of b and y Ions in Peptide MS/MS Spectra Based on Machine Learning. J Comput Commun. 2023;11(3):99-109. researchgate.net
- Fisher A, et al. Simulation of 2D electrophoresis and tandem mass spectrometry for teaching proteomics. Biochem Mol Biol Educ. 2012;40(6):400-407. (Figure 3 and associated text). researchgate.net
- De novo peptide sequencing (Wikipedia article). Wikipedia. https://en.wikipedia.org/wiki/De_novo_peptide_sequencing. Accessed Nov 2025. en.wikipedia.org