
Peptide Mapping Analysis for Reliable Research Verification
Author: Dr. Numan S. Date: July 21, 2025

What Is Peptide Mapping Analysis and Why Is It Used?
Peptide mapping analysis is an analytical technique used to confirm a protein’s identity and examine its protein sequence at the peptide level. In a typical peptide mapping workflow, a purified protein is enzymatically digested into smaller peptide fragments, which are then separated (usually by high-performance liquid chromatography) and analyzed by mass spectrometry [2]. This process generates a unique “fingerprint” of peptides that corresponds to the protein’s amino acid sequence – essentially verifying the protein’s primary structure (its amino acid sequence and any chemical modifications). Peptide mapping is widely used because it provides a high-confidence method for protein identification and structural characterization. By matching the masses or spectra of detected peptides to those expected from the known sequence, scientists can confirm that a given protein sample matches the intended sequence, or reveal differences such as mutations or modifications. This makes peptide mapping indispensable for ensuring that researchers are working with the correct protein and for maintaining quality control in biotechnological applications.
How Peptide Mapping Validates Research Outcomes
Peptide mapping plays a key role in validating experimental findings by serving as a molecular “proof of identity” for proteins under study. In research, it is often crucial to demonstrate that a purified protein or an expressed recombinant protein is exactly what it is supposed to be. Peptide mapping confirms this by providing a comprehensive check of the protein’s primary structure. For instance, if a study involves a protein engineered with a specific mutation or tag, peptide mapping will confirm the presence of that exact amino acid sequence change [4]. By verifying sequence fidelity and detecting any sequence variants, the technique validates that the protein used in experiments matches the experimental design. This prevents cases where an unexpected sequence error could lead to misleading results. In essence, peptide mapping analysis underpins the credibility of protein-related research outcomes by ensuring the protein samples have the correct identity and sequence.
In addition to confirming identity, peptide mapping supports the verification of other research outcomes such as structural or functional hypotheses. For example, if researchers suspect a protein undergoes a modification (like phosphorylation or glycosylation) in certain conditions, peptide mapping can provide evidence by identifying those modified peptides. The resulting peptide mapping data shows which peptides carry the modification and at what sites, thereby corroborating biochemical findings about protein regulation. Likewise, in experiments involving protein engineering or gene editing, peptide mapping validates outcomes by showing whether the intended protein expression changes occurred. One case study described using LC–MS peptide mapping to evaluate gene editing results: the detected peptides (and their absence or presence) confirmed whether a target protein was knocked out or an alternative protein was produced as designed [2]. In biopharma development, peptide mapping is indispensable for verifying 3rd party tested biologics, ensuring compliance with quality control standards and confirming that engineered proteins match their intended design
Step-by-Step Overview of a Peptide Mapping Workflow
A peptide mapping analysis generally follows a well-established workflow that ensures thorough characterization of a protein’s sequence. The process can be summarized in several key steps.
Sample Preparation (Denaturation and Reduction): The protein sample is first denatured (unfolded) to expose all regions of the molecule and reduced to break disulfide bonds, yielding a linear polypeptide chain. This sample preparation step is critical for allowing the enzyme full access to cleavage sites. Often, a chemical like dithiothreitol (DTT) is used for reduction, and an alkylating agent (e.g. iodoacetamide) is added to prevent disulfide reformation by modifying cysteine residues.
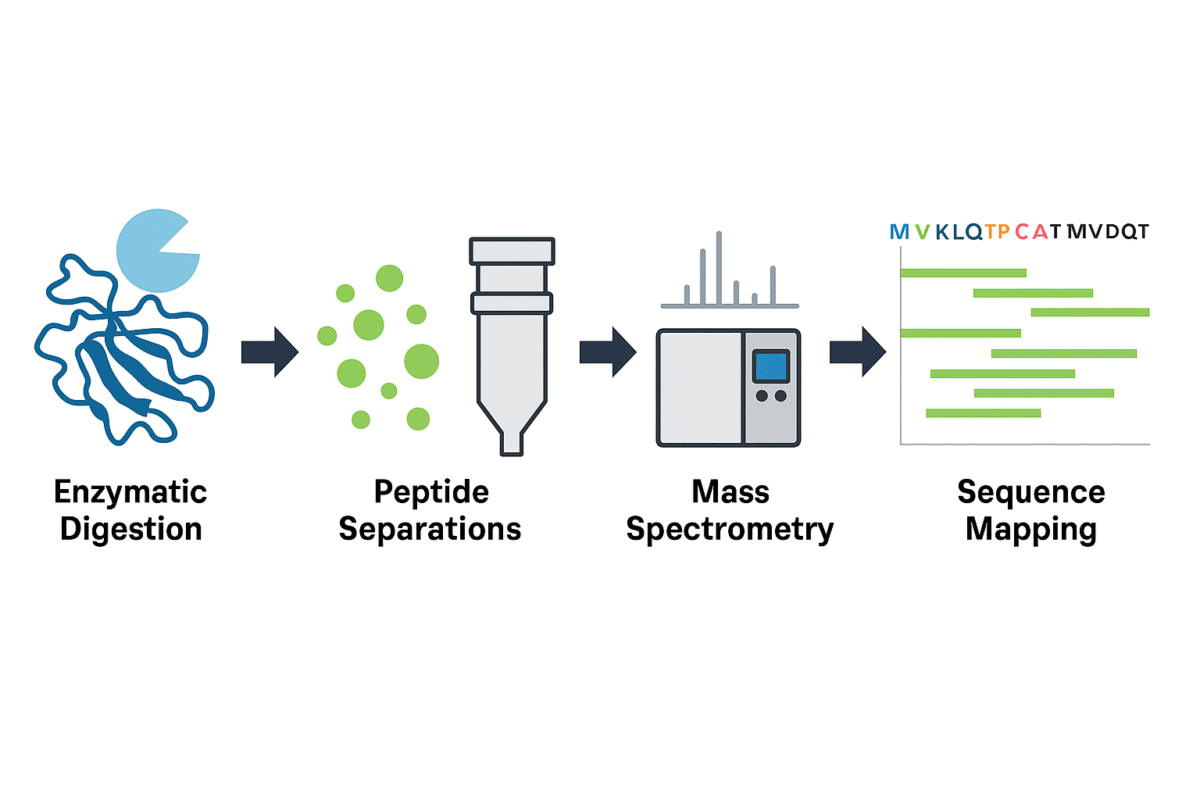
Figure 1. General workflow of peptide mapping analysis using a bottom-up approach.
- Enzymatic Digestion: The unfolded protein is then enzymatically cleaved into peptide fragments. Typically, trypsin is the enzyme of choice – it cuts at the carboxyl side of lysine or arginine residues – but other proteases (such as Lys-C, Glu-C, or chymotrypsin) may be used alone or in combination to achieve broader sequence coverage. The goal is to generate a set of overlapping peptides that together cover as much of the protein sequence as possible. Complete digestion ensures that no region of the protein escapes analysis; insufficient cleavage can leave gaps in sequence coverage, so conditions like enzyme ratio, incubation time, pH, and temperature are optimized to minimize missed cleavages. This step produces dozens to hundreds of peptides, forming the basis of the protein’s peptide “map.”
- Peptide Separations (Liquid Chromatography): The complex mixture of peptides is then separated by high-resolution liquid chromatography. Reversed-phase HPLC (RP-HPLC) is commonly used, as it can resolve peptides based on hydrophobicity, yielding a characteristic chromatographic profile with peaks corresponding to individual peptide fragments. Optimizing peptide separations is important for achieving clear results – for example, using a C18 RP column for typical tryptic peptides, or alternative methods like HILIC if certain small or polar peptides are not well retained. This chromatographic step not only simplifies the mixture for detection but also adds another dimension of data (retention time) that can help distinguish peptides.
- Detection and Mass Spectrometry Analysis: As peptides elute from the column, they are detected and analyzed. In many workflows, an LC–MS system is used, meaning a mass spectrometer acts as the detector to measure the mass-to-charge ratio of each peptide and even fragment them further (MS/MS) for sequence information. Mass spectrometry provides highly sensitive and specific detection, revealing the molecular weight of each peptide and, via tandem MS, the amino acid sequence of each peptide. In some quality control settings, UV absorbance detection may also be used (LC–UV) for a simpler peptide map if detailed identification is not required. However, MS is considered the gold standard for peptide mapping because it allows precise protein identification and modification analysis by matching peptide masses to the theoretical values from the known sequence.
- Data Analysis and Sequence Mapping: The final step is to interpret the peptide mapping data. Specialized software compares the experimentally observed peptide masses (and MS/MS spectra) with the expected peptides from the protein’s known amino acid sequence. This generates a sequence coverage map indicating which parts of the protein were observed and confirms if the protein’s primary structure matches the reference. Ideally, peptide mapping achieves high sequence coverage (often >95%), meaning almost the entire protein sequence has been verified by corresponding peptides. Any detected sequence variants or unexpected peptides are investigated at this stage, as they could indicate mutations, impurities, or modifications. The outcome is a comprehensive map of the protein’s primary structure, verifying identity and highlighting any structural features of interest.
Applications Across Research and Industry
Peptide mapping is applied across a wide range of fields in both academic research and industry, wherever detailed protein characterization is needed. In the biopharmaceutical industry, peptide mapping analysis is a cornerstone technique for developing and manufacturing protein therapeutics (such as monoclonal antibodies, enzymes, and vaccines). It is used for primary structure confirmation of biopharmaceutical products, ensuring that the protein sequence produced by a cell line matches the intended design. Regulatory guidelines (for example, ICH Q6B) actually require peptide mapping to verify the identity and primary structure of recombinant proteins as part of the quality control release process for new biologic drugs. This is because patient safety and drug efficacy depend on the protein drug having the correct amino acid sequence and no hidden modifications or sequence errors.
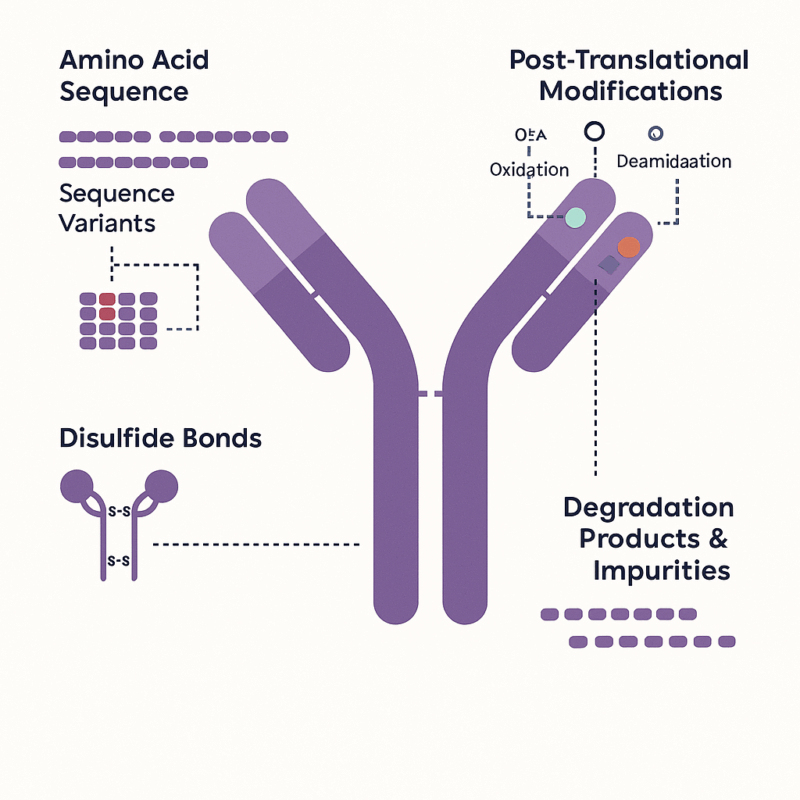
Figure 2: Example of molecular features in a therapeutic antibody that can be interrogated by peptide mapping
In academic and clinical research, peptide mapping has equally important applications. Proteomics researchers use peptide mapping (often in the form of peptide mass fingerprinting or LC-MS/MS identification) to discover and characterize proteins in complex mixtures – for example, identifying potential biomarkers or studying protein expression in cells. Here, peptide mapping assists in protein identification by matching peptide data to database sequences, which can uncover what proteins are present in a sample. Structural biologists and biochemists, on the other hand, use peptide mapping to probe specific features of proteins. For instance, mapping can locate and verify post-translational modifications that regulate protein function, or map disulfide bonds to ensure a protein is folded correctly.
Peptide mapping is also employed to detect subtle variants or impurities in research-grade proteins: if a protein preparation has a contaminant or has undergone partial degradation, the peptide map will reveal extra peptides or missing peptides corresponding to those issues. In biotechnology and genetics research, any study involving engineered proteins or mutations will benefit from peptide mapping to confirm that the engineered amino acid sequence is exactly as intended.
Even emerging fields like gene therapy use peptide mapping to verify outcomes – for example, confirming that a gene editing experiment successfully changed a protein’s sequence or expression as planned Overall, any research that demands unambiguous verification of a protein’s primary structure or comparison of protein variants can benefit from peptide mapping. Its applications span from ensuring protein identification in discovery science to serving as a high-precision QC tool in industry, underlining its versatility and importance.
Comparing Peptide Mapping to Other Protein Analysis Techniques
Peptide mapping is often considered the gold standard for detailed protein structural analysis, and it differs in significant ways from other protein characterization techniques. One common point of comparison is intact mass spectrometry versus peptide mapping. Intact protein MS involves analyzing the whole protein to get its molecular weight and perhaps some broad information, but it does not localize modifications or sequence changes. In contrast, peptide mapping (which typically uses MS on digested peptides) provides site-specific details. For example, intact MS might tell you that a protein’s mass is higher than expected (perhaps due to a modification), but peptide mapping can pinpoint which peptide (and thus which site on the protein) carries that modification[1].
This fine-grained information is critical when confirming that a protein’s amino acid sequence is correct and when characterizing PTMs. In essence, mass spectrometry is a tool often employed within peptide mapping workflows, but peptide mapping encompasses a broader strategy (digestion + separation + MS analysis + database matching) focused on sequence verification. Another technique, Edman sequencing (N-terminal sequencing), can determine the amino acid sequence of a protein directly, but it has limitations: Edman degradation can only sequence a protein up to ~30–50 residues from the N-terminus and cannot easily detect modifications at internal positions. Peptide mapping by MS/MS does not have these length limitations and can detect modifications, making it more powerful for complete protein sequence confirmation in practice.
When compared to gel-based or immunological methods (like SDS-PAGE or Western blotting), peptide mapping offers far greater precision in protein identification. SDS-PAGE can confirm a protein’s approximate size and purity, and a Western blot can confirm the presence of a protein using an antibody, but neither provides the actual sequence or the ability to detect single-residue differences. Peptide mapping, on the other hand, will reveal even a one-amino-acid change or a small PTM, because those changes alter the masses or chromatographic behavior of specific peptide fragments. This makes peptide mapping uniquely suited for confirming exact identity and detecting micro-heterogeneity (small differences between protein variants). In the context of protein’s primary structure analysis, peptide mapping surpasses other techniques by combining enzymatic specificity with the analytical power of modern MS. As a result, it is often used in tandem with or as a replacement for older methods.
Common Challenges and How to Address Them
Despite its strengths, peptide mapping is not without challenges. One common issue is achieving complete sequence coverage of the protein. Certain regions of a protein may be resistant to enzymatic digestion – for instance, highly folded domains or segments lacking the specific residues where proteases cut. If trypsin alone is used, sequences with very few lysine or arginine sites (or highly hydrophobic stretches) might not be fully cleaved, leaving large peptides that are difficult to detect or separate, ultimately resulting in gaps in coverage. Incomplete digestion can thus lead to sequence coverage below the desired level, meaning parts of the protein sequence remain unverified. To address this, researchers often optimize the digestion step: this may involve increasing digestion time, using enhanced reagents (like detergents or chaotropes to help unfold the protein), or employing multiple proteases in tandem. For example, using a second enzyme with a different cleavage specificity (such as Glu-C or chymotrypsin in addition to trypsin) can cleave regions that trypsin misses, yielding overlapping peptides that boost overall coverage. Published studies have shown that combining enzymes (e.g. trypsin and chymotrypsin) in a single workflow improved digestion efficiency and even enabled detection of subtle sequence variants that might be missed with one enzyme alone. Thus, one key to overcoming incomplete coverage is a carefully designed sample preparation and digest protocol – possibly including orthogonal enzyme digests and optimized conditions – to ensure no portion of the protein’s primary structure escapes analysis.
Another challenge in peptide mapping is the potential for sample preparation artifacts and losses. The same conditions used to reduce, alkylate, and digest proteins can sometimes induce artificial modifications (for example, deamidation or oxidation of amino acids due to high pH or prolonged exposure). These artifacts can complicate data interpretation by introducing modifications that weren’t originally present in the sample. To mitigate this, protocols are optimized to minimize exposure to harsh conditions – for instance, digesting at shorter time frames, lowering temperature, or using stabilizing additives to reduce oxidation. Automating and standardizing the prep steps (with robotic handling or commercial digest kits) can also reduce variability and inadvertent modifications. Another preparation-related challenge is peptide loss: certain peptides, especially very hydrophobic or very large ones, may stick to surfaces or precipitate, or they might elute poorly from an LC column, leading to underrepresentation. Using appropriate solvents, improving chromatographic conditions (e.g. different column chemistries or gradients), or derivatizing peptides can help recover those elusive peptides. Additionally, isobaric peptides (different sequences with the same mass) present a challenge, as they can be indistinguishable by mass alone – in these cases, using tandem MS (MS/MS) to sequence the peptides is essential to resolve their identities. Complex post-translational modifications like glycans can also make peptide spectra difficult to interpret; here, specialized approaches such as treating the sample with glycosidase enzymes or using higher-resolution MS and targeted analysis can help. Finally, data analysis can be a bottleneck if the peptide map produces a very large number of peaks. Analysts address this by using sophisticated bioinformatics tools to match peptides to the sequence and by setting acceptance criteria for identification. In summary, the common challenges in peptide mapping – incomplete digestion, peptide losses, artifactual modifications, isobaric interference, and data complexity – are managed through methodical optimizations. By tweaking enzymes, improving peptide separations, carefully controlling sample preparation conditions, and leveraging advanced MS/MS and software, scientists can largely overcome these hurdles and obtain a reliable, high-coverage peptide map.
Tools and Techniques That Enhance Peptide Mapping Precision
Continual improvements in analytical technology are enhancing the precision, speed, and sensitivity of peptide mapping. One major area of advancement is in mass spectrometry instrumentation. Modern high-resolution MS systems (such as Orbitrap or time-of-flight MS) offer greater mass accuracy and resolution, allowing more confident identification of peptides and detection of minor modifications. High mass spectrometry resolution means that peptides with nearly identical masses can be distinguished, reducing ambiguity in mapping dataemerypharma.com.
Moreover, the use of tandem MS/MS has become routine – by fragmenting each peptide ion and reading its sequence, the confidence in protein identification skyrockets, as each peptide’s amino acid sequence can be confirmed, not just its mass. Another set of tools improving precision are advanced chromatographic techniques. Ultra-high-performance LC (UHPLC) with sub-2-micron columns can achieve sharper peptide peaks and better separation, which improves the clarity of the peptide map and the quantification of modifications. Specialized column chemistries (e.g., C4 for very hydrophobic peptides, or HILIC for very polar peptides) can be employed as needed to resolve tricky segments of the proteinelementlabsolutions.com. These improvements in peptide separations directly translate to more complete maps and higher sequence coverage.
Innovations have also led to rapid peptide mapping workflows without sacrificing quality. For example, integrated systems now combine on-line digestion, fast chromatography, and ion mobility-enhanced MS to dramatically speed up the process. One report demonstrated that adding high-resolution ion mobility to an LC–MS workflow provided over 96% sequence coverage in only 20 minutes of analysis timeselectscience.net. Such rapid peptide mapping approaches are valuable for high-throughput environments or in-process monitoring, where getting quick results is important. Additionally, automation is boosting precision and reproducibility: robotic liquid handlers can perform protein digestion and sample prep in a highly consistent manner, reducing human error and variability in sample preparation.
This is especially useful in regulated labs, where an automated peptide mapping platform ensures each run is performed the same way, leading to more reliable comparisons of peptide mapping data across samples. Software tools have also become integral to enhancing precision. Dedicated peptide mapping software can automatically match peptides to the reference protein sequence, calculate % coverage, flag any sequence variants or unknown peaks (as in the multi-attribute method, MAM), and even quantify modification levels. These data processing improvements mean that even complex maps with hundreds of peptides can be interpreted quickly and consistently. Finally, the combination of multiple analytical techniques can enhance confidence: for instance, using both UV and MS detection in parallel can provide quantitative information (UV for total peptide amount) alongside structural information (MS for identity).
New techniques like top-down sequencing (analyzing the intact protein with fragmentation) are also being explored to complement bottom-up peptide mapping, providing an orthogonal confirmation of the protein’s primary structure. In conclusion, the precision of peptide mapping is being continually improved by technological advances in MS instrumentation, faster and better separations, workflow automation, and powerful data analysis software. These tools together allow researchers to obtain highly accurate, reproducible peptide maps with greater efficiency, ensuring that protein verification by peptide mapping keeps pace with the growing demands of modern biochemistry and biopharmaceutical researchemerypharma.comselectscience.net.
Frequently asked questions (FAQs) about Peptide Mapping Analysis
What are the main steps in a peptide mapping analysis?
- Peptide mapping generally involves a bottom-up proteomics approach where the protein of interest is enzymatically digested into smaller peptides, typically using trypsin. These peptides are then separated using liquid chromatography (LC) and analyzed by mass spectrometry (MS). The resulting mass spectra are matched to theoretical peptide masses or fragment patterns derived from the known protein sequence, allowing researchers to confirm sequence fidelity, detect post-translational modifications (PTMs), and identify impurities or degradation products.
Why is peptide mapping essential in confirming protein sequences?
- Peptide mapping offers high-resolution structural verification of proteins, allowing researchers to confirm the exact amino acid sequence. By comparing the experimental peptide fragments with a reference sequence, peptide mapping can detect sequence variants, truncations, or mutations that would otherwise compromise therapeutic efficacy or safety. It is especially vital for verifying the identity and integrity of recombinant proteins, therapeutic antibodies, and biosimilars.
How does peptide mapping support biologics development?
- In biologics development, peptide mapping is a core analytical method for characterizing the primary structure of protein-based drugs. It helps demonstrate batch-to-batch consistency, supports comparability studies for manufacturing changes, and enables early detection of PTMs like glycosylation or oxidation. Regulatory bodies require these data to ensure the therapeutic molecule maintains its intended structure and function throughout production and storage.
What are the regulatory expectations around peptide mapping data?
- Regulatory agencies like the FDA and EMA expect peptide mapping to be a part of the analytical characterization package for biologics. It must be performed using validated methods and provide sufficient coverage of the protein’s sequence—typically over 90%—with clear identification of any PTMs, sequence variants, or degradation products. Consistency in peptide maps across production batches is also a key requirement for demonstrating product quality and comparability.
Which tools are used to improve peptide mapping accuracy?
- Advanced analytical platforms such as high-resolution tandem mass spectrometry (HR-MS/MS), coupled with ultra-performance liquid chromatography (UPLC), greatly enhance mapping precision. Software tools for automated peptide identification, database searching, and spectral matching (e.g., Mascot, PEAKS, Byonic) are also critical. Improvements in enzyme specificity, sample preparation protocols, and hybrid methods like LC-MS/MS and ion mobility spectrometry further increase the depth and reliability of peptide mapping results.
References
- Mouchahoir T, Schiel JE. Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal Bioanal Chem. 2018;410(8):2111-2126
- Emery Pharma. Peptide Mapping: Uncovering the Protein’s Secrets. EmeryPharma website; 2023
- Rapid Novor. What is Peptide Mapping? RapidNovor website; 2022 rapidnovor.com
- Kelly LM, Renard VM. Enhancing peptide mapping sequence coverage through an automated dual protease digest. LCGC North America. 2022;40(10):520-527 chromatographyonline.com
- Element Lab Solutions. Peptide Mapping – A Beginner’s Guide. 2021