
Sterility vs Endotoxin Testing: Different Quality Safeguards
Author: Dr. Numan S. Date: January 8, 2026
Why Quality Safeguards Matter in Research and Manufacturing
Quality control safeguards are essential in both pharmaceutical manufacturing and laboratory research to ensure products and materials are safe and reliable. In industry, regulatory standards demand that sterile medicines and devices be free from both living microorganisms and pyrogenic contaminants like bacterial endotoxins. These dual requirements protect patients from infections and dangerous immune reactions. Likewise, scientists using research-grade materials rely on strict laboratory quality control tests to confirm that reagents won’t introduce microbial or endotoxin contamination that could skew experimental results.
Even minute contamination can derail an experiment or render a medical product unsafe. For example, sterility testing and endotoxin testing are two critical laboratory quality control procedures designed to catch different threats. Sterility tests verify the absence of any viable microbes, while endotoxin tests detect toxic bacterial fragments (lipopolysaccharides) that can trigger fevers or shock. Both safeguards work together to ensure that pharmaceuticals are safe for patient use and that research materials maintain the integrity of experimental outcomes. In short, a product must be not only sterile but also low in endotoxins to meet the high standards of safety and reproducibility in science and manufacturing.
What Is Sterility Testing?
Sterility testing is a quality control process used to confirm that a product or sample is free of any living microorganisms. In practice, this means exposing samples to growth media and observing them under controlled conditions – typically over a 14-day incubation period – to see if any bacteria or fungi proliferate. If no turbidity or colonies appear in the media, the sample passes as sterile; any sign of microbial growth constitutes a failure. This classical sterility test (described in USP <71>) is considered the gold standard in the pharmaceutical industry, albeit a slow one, since it requires a long incubation to ensure even low levels of contamination are detected.
The goal of sterility testing is to catch viable contaminating microbes that could spoil products or experiments. Even minimal microbial presence can alter the pH or composition of buffers and reagents, compromising their function and experimental reproducibility. Thus, sterility testing isn’t limited to finished drug vials—it’s also applied to research-grade materials like culture media, buffers, and other lab reagents to ensure they remain free of bacteria or fungi that could confound results. A preparation that passes sterility testing is considered free of live bacteria, yeast, and mold, making it safe for aseptic use or injection from a microbiological standpoint.
What Is Endotoxin Testing?
Endotoxin testing is the process of detecting and quantifying bacterial endotoxins (lipopolysaccharides from Gram-negative bacteria) in a sample to ensure it does not contain harmful levels of these pyrogens. Endotoxins are heat-stable molecules shed from bacterial cell walls, and even if bacteria are killed (for example, by sterilization), endotoxin fragments can remain and cause fever or inflammatory responses in humans and animals. Because you cannot simply “see” or filter out endotoxins the way you might bacteria, specialized assays are used to test for their presence.
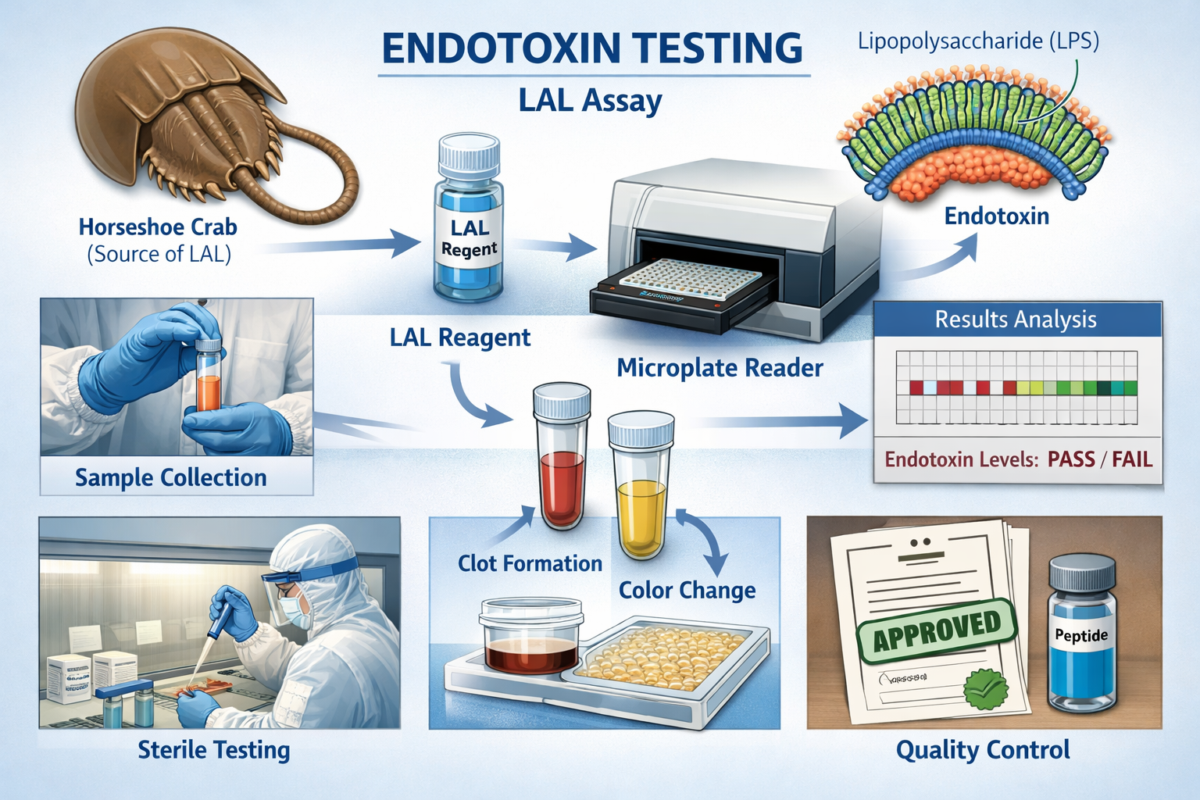
Figure 1: Endotoxin testing process explained.
The most common method is the Limulus Amebocyte Lysate (LAL) assay, which uses an enzyme from horseshoe crab blood that clots in the presence of endotoxin. In this test, a small sample of the product is mixed with the lysate; if endotoxin is present, it triggers a cascade that forms a gel or color change, indicating contamination. Modern LAL assays can be very sensitive – detecting endotoxin levels down to fractions of an EU (endotoxin unit) per milliliter – and can be performed in under an hour in formats like gel-clot, turbidimetric, or chromogenic readouts.Importantly, endotoxin testing provides a quantitative measure of pyrogen contamination. Results are typically reported in endotoxin units per milliliter (EU/mL), a standardized unit of activity. (One EU/mL corresponds roughly to 0.1–0.2 nanograms of a standard endotoxin in solution.) By measuring endotoxin levels, laboratories can confirm whether a product meets safety thresholds (for example, injectable drugs must be below a certain EU/mL limit) or if endotoxin contamination is present at levels that could affect cell cultures. In summary, endotoxin testing answers a different question than sterility testing: not “Are there live germs?” but “Could this sample contain bacterial toxins that induce fevers or confound biological systems?”.
Sterility vs Endotoxin Testing: Key Differences
Sterility testing and endotoxin testing are complementary but fundamentally different safeguards. The first key difference is what they detect: sterility tests target any viable microbial contamination (bacteria, fungi, etc.), whereas endotoxin tests target the non-living toxins that certain bacteria leave behind. An item can be sterile (no living bacteria) yet still laden with endotoxins, because common sterilization methods (heat, irradiation, filtration) may kill bacteria without destroying the endotoxins already present. Endotoxins are notoriously heat-resistant and can persist through autoclaving or gamma irradiation. This means a product that passes a sterility test is not automatically safe from a pyrogenicity standpoint. In fact, regulatory guidance emphasizes that a sterile preparation is not guaranteed to be endotoxin-free without dedicated testing. This is a critical distinction: **“sterile” and “non-pyrogenic” are separate labels that both must be verified.
A second major difference lies in testing methodology and timing. Sterility testing is a culture-based method – essentially attempting to grow any contaminating microbes over days or weeks – while endotoxin testing is a biochemical assay that yields results within an hour or two. Sterility tests often involve incubating samples in nutrient broths for 14 days, requiring careful aseptic technique and patience. By contrast, endotoxin assays like LAL are performed in vitro on small aliquots, often in microplate format, and can be automated for rapid readouts. Sterility testing tends to be qualitative (growth or no growth), whereas endotoxin testing is quantitative, measuring toxin levels in EU/mL. These differences mean that endotoxin testing can detect a risk (fever-causing LPS) that sterility testing would miss, and do so much faster. Conversely, sterility testing will catch forms of contamination (live fungi, for instance) that endotoxin tests don’t address. In practice, both tests are used in parallel to ensure comprehensive quality control: absence of living microbes and minimal endotoxin levels.
When Sterility Testing Is Required
Sterility testing is mandatory for any product that is labeled or intended to be “sterile,” especially in pharmaceuticals and medical devices. Manufacturers of injectable drugs, intravenous fluids, implantable materials, ophthalmic solutions, and other sterile dosage forms must test each lot for sterility to comply with Good Manufacturing Practices (GMP) and pharmacopeial standards. Regulatory agencies like the FDA and EMA require proof that these products contain no viable microbes, as even a single bacterial cell in a batch of injectable medicine could pose serious infection risks. Compounding pharmacies and 503B outsourcing facilities similarly must perform sterility testing on high-risk preparations to ensure patient safety.
Outside of regulated medical products, sterility testing is also crucial in research and industrial laboratory settings. Critical reagents and research-grade materials – such as cell culture media, sera, buffers, and biologics used in experiments – are often sterilized and tested to confirm the absence of contaminants. While not always “required” by law for research materials, this practice is considered essential for experimental reliability. Even a low level of microbial contamination can precipitate changes in a reagent (degrading nutrients, altering pH) that lead to erratic or false experimental results. Thus, suppliers of lab reagents typically perform sterility tests on their products, and many labs implement routine sterility checks on key solutions. In summary, sterility testing is required or expected whenever the presence of live microbes would endanger patients, compromise product shelf-life, or confound research outcomes.
Risk Management: Using Both Tests Together
Relying on sterility testing alone or endotoxin testing alone can leave blind spots; robust quality assurance requires both. Manufacturers and researchers manage risk by implementing both tests in tandem, since each addresses a different hazard. For example, a cell culture medium might be sterile-filtered to remove bacteria and fungi, but without an endotoxin test one could miss that the raw materials introduced endotoxin during production. Conversely, a sample might pass an LAL assay (low endotoxin) yet still be contaminated with live yeast or other microbes that only a sterility test would catch. Using both sterility and endotoxin testing together provides a much more complete safeguard than either alone, ensuring that a product is truly biologically clean – free of viable contaminants and of harmful bacterial residues. This dual approach is standard in pharmaceutical quality control and is increasingly adopted in high-stakes research workflows as well.
Another aspect of risk management is understanding that quality is a shared responsibility between suppliers and end-users. Reputable suppliers will perform these tests and provide documentation (COAs) attesting to sterility and low endotoxin levels, but the handling and use of materials in the lab can reintroduce risks. Both parties must do their part. For instance, a manufacturer may deliver research-grade materials that are certified endotoxin-free and sterile, but if a researcher uses poor aseptic technique or reuses non-depyrogenated containers, contamination can still occur post-delivery. Thus, laboratories are encouraged to use only endotoxin-tested reagents and practice meticulous cleanliness. As one guidance notes: it is essential to obtain reagents from trusted sources that test for endotoxins, and equally essential to use proper sterile technique and equipment cleaning to avoid introducing endotoxins during experiments. By combining supplier diligence (pre-testing products for sterility and endotoxins) with researcher diligence (maintaining sterile, pyrogen-free conditions), the risk of either live contamination or endotoxin interference is minimized.
Two Different Tests, One Shared Goal — Reliable, Safe Research
In conclusion, sterility testing and endotoxin testing are two distinct quality safeguards that serve a common goal: to ensure that scientific materials and medical products are safe, pure, and reliable. Sterility testing guards against the presence of living microbes that could cause infections or confound results, whereas endotoxin testing guards against bacterial toxins that could provoke fevers or alter biological responses. Passing one test is not a substitute for the other – a truly high-quality product or research-grade reagent must pass both, affirming that it is free of both microbial contamination and problematic endotoxin levels. Together, these tests give researchers, clinicians, and patients confidence in the integrity of the products they use.
By understanding the difference between sterility and endotoxin testing, one can better interpret quality data and make informed decisions. A common misconception is assuming that “sterile” means “endotoxin-free,” but as we’ve discussed, that’s not necessarily the case. Only by looking at both aspects can one get a full picture of product safety. Ultimately, employing both tests reflects a commitment to thorough quality control – whether in a GMP manufacturing line or an academic lab – which translates to more reliable research outcomes, safer therapies, and greater peace of mind for all stakeholders involved.
Frequently asked questions (FAQs) about Peptide Isolation
What does endotoxin testing detect that sterility testing does not?
- Endotoxin testing detects lipopolysaccharides (LPS), which are heat-stable components of the outer membrane of Gram-negative bacteria. These pyrogenic molecules can persist even after bacteria are no longer viable. Sterility testing, by contrast, evaluates whether living microorganisms are present and capable of growth under defined culture conditions. As a result, a sample may pass sterility testing yet still contain biologically active endotoxins that can interfere with sensitive assays.
When is endotoxin testing essential in research workflows?
- Endotoxin testing is essential whenever peptides or reagents are used in cell-based assays, immunological studies, in vivo models, or any application where inflammatory or cytokine responses are measured. Even trace endotoxin levels can activate immune pathways, confounding experimental outcomes. For these workflows, endotoxin testing complements sterility testing by addressing a distinct and clinically relevant contamination risk.
Why can sterilization fail to remove endotoxins?
- Common sterilization methods—such as filtration, autoclaving, or chemical disinfectants—are designed to eliminate or inactivate living microorganisms. Endotoxins, however, are not living entities and are highly resistant to heat and many chemical treatments. Once released from bacterial cell walls, endotoxins can remain in solution unless specific depyrogenation processes or validated endotoxin-control measures are applied.
How should researchers interpret sterility and endotoxin results together?
- Sterility and endotoxin results should be interpreted as complementary indicators of quality. A sterile result confirms the absence of viable microorganisms, while a low endotoxin result indicates minimal risk of pyrogen-driven biological interference. Reliable research materials typically demonstrate compliance with both criteria, particularly for applications involving biological systems. Evaluating both results together provides a more complete assessment of contamination risk.
What role do testing and handling play in overall quality assurance?
- Testing verifies the final quality of peptides and reagents, but proper handling is equally critical in maintaining that quality. Even materials that pass sterility and endotoxin testing can become compromised through improper storage, repeated transfers, or non-sterile technique. Comprehensive quality assurance therefore integrates validated testing methods with controlled manufacturing, careful handling, and appropriate storage practices to preserve experimental reliability from production through end use.
References
- Boston BioProducts. Sterility Testing | Boston BioProducts. https://www.bostonbioproducts.com/products/sterility-testing.
- FUJIFILM Wako Chemicals USA. Endotoxin: An Insidious Cell Culture Contaminant. https://www.wakopyrostar.com/endotoxin-cell-culture-contaminant.
- FUJIFILM Wako Chemicals USA. Difference Between Sterility, Bioburden, Pyrogen, and Bacterial Endotoxin Testing. https://www.wakopyrostar.com/sterility-vs-endotoxin.
- Eagle Analytical Services. Value of Sterility Testing in Pharmacy and Pharmaceuticals. https://www.eagleanalytical.com/sterility-testing-pharma.
- Eagle Analytical Services. Bacterial Endotoxins Test. https://www.eagleanalytical.com/bacterial-endotoxin-testing.
- Charles River Laboratories. Compounding Pharmacies and Bacterial Endotoxin Testing. Eureka Blog. https://www.criver.com/eureka/compounding-pharmacies-endotoxin-testing.
- Sigma-Aldrich. Cell Culture FAQs: Bacterial Endotoxin Contamination. https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/cell-culture-and-cell-culture-analysis/cell-culture-contamination/endotoxin.
- United States Pharmacopeia (USP). <71> Sterility Tests. USP 43–NF 38. Rockville, MD: United States Pharmacopeial Convention; 2020.
- U.S. Food and Drug Administration (FDA). Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers. https://www.fda.gov/media/83477/download. Published June 2012.