
The Role of Amino Acid Sequencing in Peptide Identification
Author: Dr. Numan S. Date: September 18, 2025

What Is Amino Acid Sequencing?
Amino acid sequencing is the process of determining the exact order of amino acids in a peptide or protein [6]. In other words, it reveals a molecule’s primary structure – often referred to as protein sequencing when applied to full proteins. Each peptide or protein is a chain of amino acids linked by peptide bonds, and its sequence dictates the molecule’s properties and function. Even a single change in the sequence can drastically alter a peptide’s 3D structure and biological activity, potentially leading to loss of function or disease [6]. Determining the sequence is therefore crucial for understanding what a peptide is and does.
Importantly, knowing the amino acid sequence allows researchers to predict how the peptide will fold and interact with other molecules. The sequence information is fundamental for studying molecular biology and disease mechanisms[5]. For example, the amino acid sequencing of insulin (the first protein sequenced) proved that proteins have defined sequences and laid the groundwork for modern biochemistry. Today, amino acid sequencing (also known as peptide sequencing) is an indispensable tool in peptide research, enabling scientists to characterize peptides at the most basic level and compare them across samples.
Why Sequencing Matters for Peptide Identification
Accurate sequencing is essential for peptide identification in complex biological samples. In proteomics – the large-scale study of proteins – identifying a peptide or protein among thousands of others relies on knowing its unique amino acid sequence [1]. Sequence data act like a molecular fingerprint. By determining a peptide’s sequence, scientists can match it to known proteins or confirm it as a novel entity. Thus, amino acid sequencing underpins most mass spectrometry–based protein identification workflows in proteomics, which aim to catalog all proteins in a cell or tissue.
Sequencing also tells us exactly which peptide we have, ensuring that researchers are studying the correct molecule. This is especially important because many peptides may be similar in composition but differ by a critical amino acid. Peptide identification by sequencing can reveal post-translational modifications (like phosphorylation or glycosylation) that standard protein assays might miss [6]. Such modifications can change a peptide’s identity and function. Moreover, knowing the precise sequence can uncover disease-related mutations – for instance, a single amino acid substitution in a protein that causes a genetic disorder[6]. In short, amino acid sequencing provides definitive identification of peptides, which is vital for everything from basic biochemistry to clinical diagnostics.
Techniques for Amino Acid Sequencing
Edman degradation is a pioneering sequencing technique developed in the 1950s. It systematically removes one amino acid at a time from the N-terminus of a peptide and identifies each residue. In each cycle of Edman degradation, a reagent (phenyl isothiocyanate) reacts with the peptide’s N-terminal amino acid, which is then cleaved and analyzed, revealing that amino acid’s identity. This method provides the sequence in order, one residue after another. Edman degradation is highly accurate for short peptides (up to around 30–50 amino acids) and was instrumental in early protein sequencing efforts. However, it is labor-intensive and time-consuming – each amino acid removal/analysis cycle can take close to an hour[5]. Edman sequencing also requires a relatively pure peptide and a free (unmodified) N-terminus. Many proteins have a blocked N-terminus (e.g. an acetylation on the first amino acid), which Edman chemistry cannot handle[5]. These limitations mean Edman degradation is now mostly used for small, purified peptides or for confirming the N-terminal sequence of a protein, rather than high-throughput peptide identification.
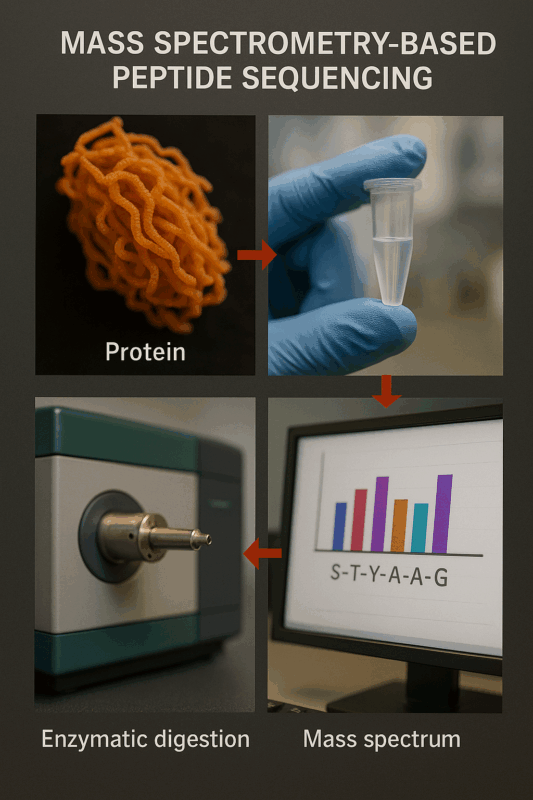
Figure 1: Example workflow of mass spectrometry-based peptide sequencing
Modern peptide sequencing is dominated by mass spectrometry techniques. In mass spectrometry-based protein sequencing, proteins are first broken into smaller peptide fragments (typically 5–30 amino acids long) using specific proteases (e.g. trypsin). These peptide fragments are then ionized and introduced into the mass spectrometer, which measures the mass-to-charge (m/z) ratios of the ions. In tandem MS (MS/MS) sequencing, peptide ions are further fragmented into smaller pieces inside the instrument. The pattern of fragment masses is essentially a puzzle that can be interpreted to determine the peptide’s amino acid sequence[6]. By analyzing the spectrum of fragments (often with sophisticated software), researchers infer the sequence of the peptide one amino acid at a time. Mass spectrometry is extremely sensitive and fast, capable of sequencing peptides in complex mixtures and identifying multiple peptides simultaneously. These sequencing techniques have revolutionized proteomics by allowing high-throughput peptide identification even in very complex samples[6][5]. Different MS-based methods exist – for example, de novo sequencing, which deduces sequences without any database, and database-driven sequencing, which matches spectra to known sequences. Together, Edman degradation and mass spectrometry form the core toolbox of amino acid sequencing, each with its niche applications.
Applications of Amino Acid Sequencing in Research
Amino acid sequencing has broad applications across biological and medical research. In proteomics research, sequencing allows scientists to catalog and quantify proteins in cells and tissues. By sequencing peptides obtained from a sample, researchers can identify which proteins are present (and in what amounts) under different conditions[6]. This is fundamental for understanding cellular biology – for example, comparing the proteome of a healthy cell vs. a diseased cell to find proteins that change. Sequencing is also critical for discovering biomarkers: unique peptide or protein sequences that signify a disease state. In biomarker discovery studies, mass spectrometry sequencing is used to hunt for peptide fragments in blood or tissue samples that might serve as early indicators of cancer, cardiovascular disease, and other conditions[6]. Without the ability to sequence and identify these peptides, such protein-based diagnostics would not be possible.
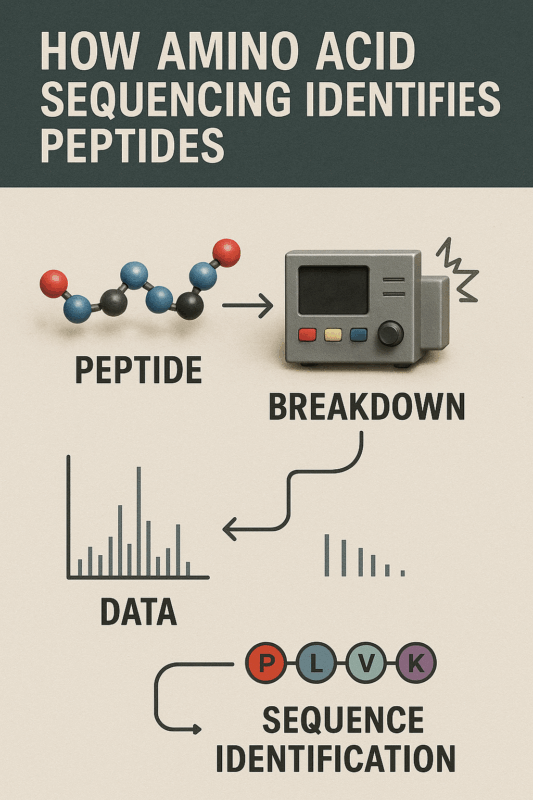
Figure 2: Step-by-step visual explaining how amino acid sequencing identifies peptides
In pharmaceutical and biotech research, amino acid sequencing plays a key role in drug development. Many modern drugs are biologics – either proteins (like antibodies) or peptides – and verifying their amino acid sequence is crucial for ensuring the drug is correct and pure. For instance, if scientists design a therapeutic peptide, they will use sequencing techniques to confirm that the manufactured product has the exact intended sequence. Sequencing is also used to identify bioactive peptides in nature that could inspire new drugs. A classic example is the discovery of peptide hormones and neurotransmitters, whose sequences had to be determined to understand their function. In drug discovery, researchers routinely sequence peptides to find those that can bind to disease-related targets or to improve a peptide’s properties. Identifying the sequence of peptides from venom, bacteria, or other organisms has led to new antibiotics and anti-cancer compounds. Additionally, sequencing helps in identifying protein targets of drugs: scientists may sequence proteins that bind a drug to see exactly what the drug is interacting with[6]. In summary, from mapping the human proteome to engineering novel therapeutics, amino acid sequencing is a driving force in both basic research and applied biomedical science.
Challenges in Amino Acid Sequencing
Despite its importance, amino acid sequencing can face significant challenges, and different sequencing techniques have their own limitations. For classical Edman degradation, one major challenge is efficiency and throughput. The method cannot easily scale up to sequence many peptides at once because it must be done stepwise for each peptide. It requires a substantial amount of sample (often in the picomole range per sequence) and becomes error-prone or fails outright for longer peptides. Practically, Edman degradation is reliable only for sequences under about 30 amino acids[5]. Another limitation is that any modification blocking the peptide’s N-terminus (such as N-terminal acetylation, common in eukaryotic proteins) will prevent the Edman reaction from proceeding[5]. Since a large fraction of proteins have blocked N-termini, Edman sequencing often can’t be applied to them without special preparation. These factors make Edman degradation slow and limited in scope, which is why it has largely been superseded by faster methods in many settings.
Mass spectrometry-based peptide identification also has its hurdles. A typical proteomic sample – say a cell lysate – contains thousands of different peptides, yielding extremely complex spectra.. This complexity can make it hard to detect low-abundance peptides, which may get overshadowed by more abundant signals.. Additionally, some amino acids produce nearly identical fragment ions in MS/MS. For example, leucine and isoleucine are isomers with the same mass, so standard mass spectrometry cannot distinguish between them directly.. Specialized approaches or additional chemical derivatization are needed to tell them apart. Another challenge is that bottom-up proteomic sequencing (digesting proteins into peptides first) relies on databases to identify proteins – the fragment patterns are typically matched to sequences in a protein database. If the organism’s genome is not well annotated or the protein is completely novel, database matching may fail[5]. In such cases, researchers must attempt de novo sequencing, which is more difficult and not always accurate for every peptide. Furthermore, interpreting mass spectra requires advanced bioinformatics tools and expertise. There is a risk of misidentification – for instance, assigning spectra to the wrong peptide, especially if a reference database is incomplete[5]. Finally, certain post-translational modifications can complicate sequencing; while MS can detect modifications, modified peptides may ionize or fragment differently, requiring extra steps (like targeted enrichment or specific fragmentation techniques) to confidently sequence them[5].. Ongoing improvements in instrumentation and analysis algorithms continue to address these challenges, making peptide sequencing more robust even for very complex samples.
Conclusion: The Critical Role of Sequencing in Peptide Identification
In conclusion, amino acid sequencing plays a critical role in peptide identification and is fundamental to modern bioscience. By determining the precise sequence of amino acids, researchers can identify what a peptide is, understand its function, and distinguish it from similar molecules. Classical methods like Edman degradation paved the way by confirming that proteins have defined sequences, while modern mass spectrometry-based approaches now enable high-throughput identification of peptides in extremely complex mixtures. Together, these tools ensure that scientists can decode the information contained in proteins and peptides. This capability is the backbone of proteomics research, which relies on sequencing data to map out the protein landscape of cells and organisms. It also underlies many advances in medicine – from the design of peptide drugs to the discovery of new disease biomarkers – where knowing the exact amino acid sequence is paramount.
As the field progresses, with emerging sequencing techniques and technologies, the process of peptide and protein sequencing is becoming faster, more sensitive, and more comprehensive. Challenges remain, but ongoing innovations are continually improving accuracy and throughput. Amino acid sequencing will remain indispensable for peptide research and protein science, driving discoveries in biology and helping to translate those discoveries into real-world applications. In essence, sequencing is to peptides what decoding is to a message – it reveals the information necessary to understand and utilize these building blocks of life. By unravelling peptide sequences, scientists can identify nature’s peptides and engineer new ones, solidifying the central role of sequencing in both the identification and the innovation of peptides.
Frequently asked questions (FAQs) about Amino Acid Sequencing in Peptide Identification
What is amino acid sequencing and how is it performed?
- Amino acid sequencing is the analytical process of determining the precise order of amino acids in a peptide or protein chain. It is performed using classical chemical methods such as Edman degradation, which sequentially removes and identifies N-terminal residues, or modern mass spectrometry-based techniques (e.g., tandem MS/MS) that analyze fragment ion patterns to reconstruct the peptide sequence.
Why is amino acid sequencing essential for peptide identification?
- Sequencing allows researchers to confirm a peptide’s identity, detect post-translational modifications, and verify synthesis accuracy. It ensures that experimental peptides match theoretical compositions, a critical factor for reproducibility, drug development, and biological research integrity.
What techniques are most commonly used in sequencing?
- The two most common approaches are Edman degradation and mass spectrometry (MS). While Edman degradation provides high accuracy for short peptides, MS-based sequencing offers rapid, high-throughput analysis of complex peptide mixtures, making it the dominant method in proteomics.
What challenges do researchers face with amino acid sequencing?
- Difficulties include incomplete N-terminal labeling, peptide degradation, and the presence of chemical modifications that obscure mass spectra. Additionally, complex mixtures and low peptide abundance can limit detection sensitivity, requiring advanced sample preparation and computational analysis.
How will sequencing evolve with AI and new technology?
- Emerging AI-driven algorithms and deep-learning models are transforming peptide sequencing by improving spectral interpretation, predicting fragmentation patterns, and automating identification workflows. Integration of machine learning with high-resolution MS data promises faster, more accurate sequencing for biomedical and pharmaceutical research.
References
- Noor Z, Ahn SB, Baker MS, Ranganathan S, Mohamedali A. Mass spectrometry-based protein identification in proteomics – a review. Brief Bioinform. 2021;22(2):1620-1638pubmed.ncbi.nlm.nih.gov
- Bishop BM, et al. Discovery of Novel Antimicrobial Peptides from Varanus komodoensis (Komodo Dragon) by Large Scale Analyses and De Novo–Assisted Sequencing using Electron Transfer Dissociation Mass Spectrometry. J Proteome Res. 2017;16(3):1470-1482sci.news
- Alfaro JA, et al. The emerging landscape of single-molecule protein sequencing technologies. Nat Methods. 2021;18(6):604-617nature.com
- Hughes C, Ma B, Lajoie GA. De novo sequencing methods in proteomics. Methods Mol Biol. 2010;604:105-121rapidnovor.com
- Edman P. A method for the determination of amino acid sequence in peptides. Arch Biochem. 1949;22:475rapidnovor.com
- JPT Peptide Technologies. Peptide Sequencing: Techniques and Applications. (Blog Article) 2021jpt.com